журнал учета работы изделия к формуляру медицинского изделия
Журнал учета работы изделия к формуляру медицинского изделия
от 19 января 2017 года N 11н
(с изменениями на 20 ноября 2020 года)
Документ с изменениями, внесенными:
приказом Минздрава России от 22 апреля 2019 года N 239н (Официальный интернет-портал правовой информации www.pravo.gov.ru, 26.06.2019, N 0001201906260037);
приказом Минздрава России от 20 ноября 2020 года N 1236н (Официальный интернет-портал правовой информации www.pravo.gov.ru, 21.12.2020, N 0001202012210123) (вступил в силу с 1 января 2021 года).
— Примечание изготовителя базы данных.
В соответствии со статьей 38 Федерального закона от 21 ноября 2011 г. N 323-ФЗ «Об основах охраны здоровья граждан в Российской Федерации» (Собрание законодательства Российской Федерации, 2011, N 48, ст.6724; 2013, N 48, ст.6165; 2015, N 1, ст.85; N 27, ст.3951) и подпунктом 5.2.192_1 Положения о Министерстве здравоохранения Российской Федерации, утвержденного постановлением Правительства Российской Федерации от 19 июня 2012 г. N 608 (Собрание законодательства Российской Федерации, 2012, N 26, ст.3526; 2013, N 16, ст.1970; N 20, ст.2477; N 22, ст.2812; N 33, ст.4386; N 45, ст.5822; 2014, N 12, ст.1296; N 37, ст.4969; 2015, N 2, ст.491; N 12, ст.1763; 2015, N 23, ст.3333; 2016, N 2, ст.325; N 9, ст.1268; N 27, ст.4497; N 28, ст.4741; N 34, ст.5255; N 49, ст.6922),
2. Установить, что утвержденные пунктом 1 настоящего приказа требования применяются к технической и эксплуатационной документации производителей (изготовителей) медицинских изделий, заявления о государственной регистрации которых представлены в Федеральную службу по надзору в сфере здравоохранения после вступления в силу настоящего приказа.
в Министерстве юстиции
10 марта 2017 года,
регистрационный N 45896
УТВЕРЖДЕНЫ
приказом
Министерства здравоохранения
Российской Федерации
от 19 января 2017 года N 11н
Требования к содержанию технической и эксплуатационной документации производителя (изготовителя) медицинского изделия
(с изменениями на 20 ноября 2020 года)
I. Общие положения
1. Настоящие Требования определяют перечень информации, подлежащей указанию в технической и эксплуатационной документации производителя (изготовителя) медицинского изделия.
2. Производитель (изготовитель) медицинского изделия разрабатывает техническую и (или) эксплуатационную документацию, в соответствии с которой осуществляются производство, изготовление, хранение, транспортировка, монтаж, наладка, применение, эксплуатация, в том числе техническое обслуживание, а также ремонт, утилизация или уничтожение медицинского изделия.
3. Настоящие Требования не распространяются на медицинские изделия, которые изготовлены по индивидуальным заказам пациентов, к которым предъявляются специальные требования по назначению медицинских работников и которые предназначены исключительно для личного использования конкретным пациентом, а также медицинские изделия, предназначенные для использования на территории международного медицинского кластера.
II. Требования к содержанию технической документации производителя (изготовителя) на медицинское изделие, за исключением программного обеспечения, являющегося медицинским изделием, в том числе программного обеспечения с применением технологий искусственного интеллекта
1) наименование медицинского изделия, иную информацию, позволяющую идентифицировать медицинское изделие, например, номер модели, варианты модификаций (исполнений) медицинского изделия;
2) назначение медицинского изделия и принципы действия;
3) показания и противопоказания к применению медицинского изделия;
4) информацию о потенциальных потребителях медицинского изделия;
5) описание основных функциональных элементов медицинского изделия, которое может сопровождаться схемами, фотографическими изображениями, рисунками, диаграммами и иными пояснениями;
6) описание составных частей (узлов) медицинского изделия (при наличии);
7) описание принадлежностей, медицинских изделий или изделий, не являющихся медицинскими, но предусмотренных для использования в комбинации с заявленным медицинским изделием (при наличии);
8) перечень и описание материалов медицинского изделия, вступающих в непосредственный или опосредованный контакт с организмом пациента (телом человека);
9) данные о маркировке медицинского изделия и его упаковке;
10) перечень рисков, идентифицированных в процессе анализа риска, и описание способов управления этими рисками в целях снижения их до допустимого уровня (при наличии);
11) сведения о верификации и валидации медицинского изделия, которые использовались для доказательства соответствия медицинского изделия установленным требованиям, в том числе результаты:
а) испытаний в испытательных лабораториях (центрах);
б) лабораторных и (или) заводских испытаний, включая результаты испытаний в условиях, имитирующих эксплуатационные;
в) лабораторных испытаний на животных для подтверждения правильности концепции готового медицинского изделия;
12) перечень материалов животного и (или) человеческого происхождения с указанием сведений об их биологической совместимости и безопасности, о выборе источников (доноров), взятии проб, обработке, хранении и обращении с данными материалами (при наличии);
13) информацию о проведенных испытаниях, протоколах испытаний, анализе полученных данных;
14) ссылки на предыдущие модификации медицинского изделия или подобные модификации медицинских изделий, находящихся в обращении, в случае использования в технической документации информации о подобных или предыдущих модификациях медицинского изделия для доказательства соответствия медицинского изделия требованиям безопасности и эффективности;
15) информацию об основных стадиях проектирования медицинского изделия и производственных процессах, которая может сопровождаться схемами, фотографическими изображениями, рисунками, диаграммами и иными пояснениями;
16) сведения о документах, подтверждающих качество лекарственного препарата, фармацевтической субстанции, биологического материала и иного вещества, с использованием которых оно произведено или которые входят в состав медицинского изделия и которые предназначены для применения только с учетом назначения медицинского изделия, определенного производителем, и выданных в соответствии с законодательством страны происхождения лекарственного препарата, фармацевтической субстанции, биологического материала и иного вещества;
17) описание метода стерилизации, сведения о методах валидации в отношении процесса стерилизации (включая испытания на биологическую нагрузку, наличие пирогенных веществ, наличие остаточного количества стерилизующего вещества) и о валидации процесса упаковывания (в случае, если медицинское изделие поставляется в стерильном виде);
18) информацию о процессе проектирования, разработки и валидации программного обеспечения, используемого в готовом медицинском изделии (в случае наличия в медицинском изделии программного обеспечения, обеспечивающего его правильную эксплуатацию и (или) применение по назначению);
19) требования к техническому обслуживанию и ремонту медицинского изделия;
20) порядок и условия утилизации или уничтожения медицинского изделия.
5. Техническая документация медицинского изделия для диагностики in vitro, помимо информации, указанной в пункте 4 настоящих Требований, должна содержать:
1) описание назначения медицинского изделия, включая:
а) описание целевого аналита, сведения о его научной обоснованности, указание на качественный, полуколичественный или количественный вид аналита;
б) функциональное назначение (например, скрининг, мониторинг, диагностика или вспомогательное средство в диагностике);
в) специфическую патологию, состояние или фактор риска, для обнаружения, определения или дифференцирования которого предназначено медицинское изделие для диагностики in vitro;
г) тип анализируемого образца;
д) популяционные, демографические аспекты применения медицинского изделия;
2) конкретизацию профессионального уровня потенциальных пользователей (например, врач клинической лабораторной диагностики, медицинский лабораторный техник (фельдшер-лаборант), иной специалист);
4) описание условий транспортировки;
5) сведения об аналитической чувствительности (порог обнаружения), аналитической специфичности, диагностической чувствительности и диагностической специфичности;
6) описание измерительных процедур, метрологической прослеживаемости значений калибраторов и контрольных материалов;
7) данные по стабильности медицинского изделия, подтверждающие заявленные срок годности, стабильность при применении и стабильность при транспортировке.
II_1. Требования к содержанию технической документации производителя (изготовителя) на программное обеспечение, являющееся медицинским изделием
(Глава дополнительно включена с 1 января 2021 года приказом Минздрава России от 20 ноября 2020 года N 1236н)
1) наименование программного обеспечения, являющегося медицинским изделием, и иную информацию, позволяющую идентифицировать программное обеспечение, являющееся медицинским изделием, например, варианты исполнений, версию программного обеспечения, являющегося медицинским изделием.
Техническая документация на программное обеспечение должна содержать в том числе разъяснение порядка нумераций его версий;
2) сведения о назначении программного обеспечения, являющегося медицинским изделием, и принципах его действия;
3) информацию о потенциальных потребителях (пользователях) программного обеспечения, являющегося медицинским изделием;
4) сведения о функции интерпретации, источнике набора данных, аппаратной платформе, способе размещения программного обеспечения, являющегося медицинским изделием, и предоставлении доступа к нему;
Приказ Министерства здравоохранения Российской Федерации от 6 июня 2012 г. N 4н «Об утверждении номенклатурной классификации медицинских изделий» (зарегистрирован Министерством юстиции Российской Федерации 9 июля 2012 г., регистрационный N 24852), с изменениями, внесенными приказами Министерства здравоохранения Российской Федерации от 25 сентября 2014 г. N 557н (зарегистрирован Министерством юстиции Российской Федерации 17 декабря 2014 г., регистрационный номер N 35201) и от 7 июля 2020 г. N 686н (зарегистрирован Министерством юстиции Российской Федерации 10 августа 2020 г., регистрационный номер N 59225).
6) сведения о наличии (отсутствии) в программном обеспечении, являющемся медицинским изделием, технологий искусственного интеллекта, и их описание;
7) описание составных частей, модулей, блоков программного обеспечения, являющегося медицинским изделием, которое может сопровождаться структурными схемами архитектуры программного обеспечения;
8) информацию о возможных изменениях программного обеспечения, являющегося медицинским изделием, которые влияют (не влияют) на неизменность его функционального назначения и (или) принципа действия (при наличии);
9) информацию о способе получения пользователем сведений о текущей версии программного обеспечения, являющегося медицинским изделием, и порядке его обновления;
10) характеристики принадлежностей программного обеспечения, являющегося медицинским изделием, медицинских изделий или изделий, не являющихся медицинскими, но предусмотренных для использования в комбинации с программным обеспечением, являющимся медицинским изделием, а также описание специального оборудования и (или) программного обеспечения, тестовых баз данных, разработанных производителем (изготовителем) для использования программного обеспечения, являющегося медицинским изделием (при наличии);
Журнал учета работы изделия к формуляру медицинского изделия
Единая система программной документации
Требования к содержанию и оформлению
Unified system for program documentation. Technical and operation data card. Requirements for contents and form of presentation
Дата введения 1980-01-01
Постановлением Государственного комитета CCCР по стандартам от 18 декабря 1978 г. N 3351 дата введения установлена 01.01.80
ПЕРЕИЗДАНИЕ. Январь 2010 г.
Настоящий стандарт устанавливает правила составления программного документа «Формуляр», определенного ГОСТ 19.101-77.
1. ОБЩИЕ ПОЛОЖЕНИЯ
1.1. Структура и оформление документа устанавливаются в соответствии с ГОСТ 19.105-78.
Информационную часть (аннотацию и содержание) допускается в документ не включать.
1.2. В основную часть документа должны входить следующие разделы:
периодический контроль основных характеристик при эксплуатации и хранении;
свидетельство о приемке;
свидетельство об упаковке и маркировке;
сведения о рекламациях;
сведения о хранении;
сведения о закреплении программного изделия при эксплуатации;
сведения об изменениях;
Состав и содержание разделов документа определяют в соответствии с особенностями программных изделий.
При необходимости допускается дополнять документ другими разделами или объединять отдельные разделы, а также помещать их в приложениях.
2. СОДЕРЖАНИЕ РАЗДЕЛОВ
2.1. В разделе «Общие указания» приводят общие указания для обслуживающего персонала по эксплуатации программного изделия, заполнению и ведению его формуляра, например:
«Перед эксплуатацией необходимо внимательно ознакомиться с соответствующими эксплуатационными документами (приводятся наименования документов).
Формуляр должен находиться в подразделении, ответственном за эксплуатацию программного изделия».
2.2. В разделе «Общие сведения» указывают наименование программного изделия, его обозначение, наименование предприятия-изготовителя, номер программного изделия предприятия и другие общие сведения о программном изделии.
2.3. В разделе «Основные характеристики» приводят необходимые при эксплуатации программного изделия значения основных характеристик (например, функциональных, надежности и др.).
2.4. В разделе «Комплектность» перечисляют все непосредственно входящие в программное изделие другие программные изделия и документацию в соответствии с комплектностью, указанной в технических условиях на программное изделие.
При наличии ведомости эксплуатационных документов в формуляре делают на нее ссылку без перечисления эксплуатационных документов (форма 1 приложения).
2.5. В разделе «Периодический контроль основных характеристик при эксплуатации и хранении» указывают наименование измерения проверяемых характеристик, требуемую периодичность контроля (форма 2 приложения).
Фактические значения характеристик записывают в раздел формуляра после каждого определения.
2.6. В разделе «Свидетельство о приемке» приводят свидетельство о приемке программного изделия, подписанное лицами, ответственными за приемку (форма 3 приложения).
2.7. В разделе «Свидетельство об упаковке и маркировке» помещают сведения об упаковке программного изделия, подписанные лицами, ответственными за упаковку (форма 4 приложения).
2.8. В разделе «Гарантийные обязательства» приводят гарантийные обязательства предприятия-изготовителя.
2.9. В разделе «Сведения о рекламациях» приводят краткое изложение порядка предъявления рекламации и регистрируют все предъявленные рекламации, их содержание и принятые меры (форма 5 приложения).
2.10. В разделе «Сведения о хранении» указывают сроки хранения и условия хранения программного изделия (форма 6 приложения).
2.11. В разделе «Сведения о закреплении программного изделия при эксплуатации» указывают фамилии и должности лиц, за которыми закрепляют программное изделие (форма 7 приложения).
2.12. В разделе «Сведения об изменениях» указывают основание для внесения изменений, содержание изменения с указанием его порядкового номера, а также должность, фамилию и подпись лица, ответственного за проведение изменения (форма 8 приложения).
2.13. В разделе «Особые отметки» оставляют несколько чистых листов для специальных отметок, которые вносят во время эксплуатации программного изделия.
2.14. В качестве приложений к формуляру могут быть справочные материалы и дополнительные документы (например, журнал учета работы), необходимые при эксплуатации программного изделия.
Журнал технического обслуживания медицинской техники и оборудования
Наличие журнала обязательное требование для любого медучреждения. В нем:
Журнал ТО всегда должен храниться в общем доступе: на посту медсестры, у управляющего / заведующего клиникой или медцентром, в кабинете заведующего отделением, главного инженера / техника и т.д.
Качественное техническое обслуживание медицинского оборудования в Cordismed в соответствии со всеми актуальными регламентами и нормативной базой.
Правильное оформление журнала ТО, договора, приложений. Полное документальное и техническое сопровождение.
Свяжитесь с нами: ответим на любые вопросы по оборудованию. Поможем проверить текущее состояние. Проведем совместную дистанционную диагностику. Или приедем для полноценной проверки на месте.
Почему журналах технического обслуживания важен на самом деле
Кто должен заполнять журнал по техническому обслуживанию, кто отвечает за журнал
Ответственность за журнал обслуживания со стороны клиники
Отвечает за журнал сотрудник клиники, ответственный по должностной инструкции за эксплуатацию медицинского оборудования :
Соответственно, такому сотруднику необходимо следить за наличием журнала, его сохранностью и внешним видом, правильностью заполнения и полной фиксацией всех действий с медтехникой.
При этом вы всегда можете рассчитывать на помощь нашего сервисного куратора и инженеров, работающих с техникой со стороны сервисной компании.
У нас большой опыт в ведении подобной документации, и мы всегда рады помочь вам. Для клиник на обслуживании Кордисмед мы всегда берем комплексное ведение технической документации на себя, гарантируем правильность ее заполнения.
Ответственность сервисной компании за журнал технического обслуживания
За правильность заполнения и актуальные данные в журнале отвечает именно инженер или техник, работающий в данный момент с оборудованием. Контролирует правильность заполнения сервисный куратор, который также является сотрудником обслуживающей компании. Другими словами:
Инженеры и сотрудники сервисного центра отвечают за правильность и своевременность заполнения журнала ТО медицинской техники, за актуальность информации в нем.
Пример (образец) журнала технического обслуживания медицинской техники
Как выглядит журнал ТО
Где взять образец журнала ТО
При заключении договора на обслуживание мы предоставляем журнал со всеми необходимыми разделами вместе с пакетом документов, то есть вам не нужно искать и покупать его самостоятельно.
В целом, есть несколько вариантов получения журнала
Скачать журнал технического обслуживания медицинского оборудования
Как заполнять журнал технического обслуживания и ремонта медицинского оборудования. Разделы журнала
Независимо от внешнего вида и особенностей оформления, любой журнал ТО, как правило. содержит следующие разделы:
Дополнительно в журнале могут быть указаны:
Также эти разделы могут отсутствовать или быть вынесены в отдельные пункты / приложения к договору на техническое обслуживание.
Список оборудования включает в себя перечень аппаратов, которые обслуживаются в данной сервисной компании по данному договору.
Важно, что в данном разделе может быть перечислено далеко не все оборудование больницы / отделения, а только то, которое фигурирует в договоре на сервисное обслуживание.
Например, в одной поликлинике рентгеновское, лабораторное, и все остальное оборудование могут обслуживать разные компании. В таком случае, в ЛПУ будет три журнала обслуживания, каждый со своим списком техники, работами и т.д.
Раздел заполняется инженером или куратором по мере выполнения работ.
Инструктаж по эксплуатации медицинского оборудования для персонала в III разделе журнала заполняется куратором при прохождении сотрудниками ЛПУ соответствующих инструктажей. Как правило, это: электробезопасность. общая техника безопасности, особенности эксплуатации оборудования.
Сведения о неисправностях в III разделе журнала медтехники вносятся инженером при обнаружении каких-либо дефектов в процессе технического обслуживания. Здесь также содержатся данные об их устранении, замененных деталях и так далее.
Пример внешнего вида разделов с регламентом ТО и графиком плановых выездов инженеров находится в шаблоне журнала технического обслуживания и ремонт медицинской техники, который можно скачать по ссылке в этой статье.
Если вам нужна помощь с обслуживанием или ремонтом медицинского оборудования, мы всегда готовы оперативно устранить любые неисправности, выполнить профилактические работы, и возьмем на себя все технические и организационные моменты работ, в том числе и полное оформление документов, журнала ТО.
Техническая и эксплуатационная документация на медицинские изделия
Наличие и технической и эксплуатационной документации на медицинское изделие (далее- МИ/медизделие) является необходимым условием для государственной регистрации и формирования регистрационного досье.
Далее рассказываем о том, какая документация на медизделия существует и какие требования к ней предъявляет действующее законодательство. Соблюдать требования важно, так как это является необходимым условием государственной регистрации медицинского изделия в рамках национальной системы.
Документация на медицинские изделия
Действующее законодательство РФ предусматривает следующие документы на медизделия:
Требования к документации на медизделие
Требования не распространяются на медизделия, которые:
Требования к содержанию технической документации на медицинское изделие
Техническая документация, представляемая в составе регистрационного досье на медицинское изделие, должна содержать:
Требования к содержанию эксплуатационной документации производителя (изготовителя) медицинского изделия
В состав регистрационного досье помимо технической документации входит и эксплуатационная. Эксплуатационная документация должна содержать:
Техническая и эксплуатационная документация медицинского изделия для диагностики in vitro
Документация на медизделия для диагностики in vitro должна соответствовать всем указанным выше требованиям и дополнительно к ним отвечать следующим параметрам.
Техническая документация должна включать в себя:
Эксплуатационная документация должна содержать:
Форма документации на медизделие
Эксплуатационная документация предоставляется производителем (изготовителем) для ознакомления потребителю на бумажном носителе (вместе с медизделием или отдельно от него) и в форме электронного документа, доступного в Интернете или на экране, являющемся частью медизделия.
В отношении медицинских изделий 1 и 2а классов потенциального риска их применения потребителю для ознакомления может быть предоставлена эксплуатационная документация в сокращенном виде, при условии, что объем предоставляемой информации достаточен для его применения по назначению и такое применение безопасно.
Наша помощь в регистрации медицинских изделий
Факультет медицинского права оказывает следующий спектр услуг по сопровождению процесса регистрации медицинских изделий в Росздравнадзоре (как в комплексе, так и выборочно):
Как упростить ведение формуляра на медицинское оборудование?

В данной статье мы расскажем:
Что такое формуляр на медицинское оборудование?
Формуляром на оборудование называется документ, который содержит информацию, удостоверяющую гарантии производителя, основные свойства и параметры изделия, сведения о его техническом состоянии, сертификации, утилизации, а также данные, вносимые в период эксплуатации: условия и длительность функционирования, техобслуживание, ремонт и другие сведения.
Формуляры должны быть приложены производителем медицинского оборудования при поставке, такие требования содержатся в письме Минздрава РФ № 293-22/233 от 27.10.2003 года. В случае отсутствия формуляра медицинского оборудования, ЛПУ может разработать его самостоятельно.
Какие разделы должен содержать формуляр?
Формуляр на медицинское оборудование представляет собой пакет документов, содержащих данные об изделии в соответствии с паспортом изделия и эксплуатационной документацией.
Согласно ГОСТа 2.610-2006 (раздел 7) формуляр должен иметь:
Раздел «Общие указания»
Раздел «Основные сведения об изделии»
Раздел «Основные технические данные»
Раздел «Индивидуальные особенности изделия»
Раздел «Ресурсы, сроки службы и хранения, гарантии изготовителя (поставщика)»
Раздел «Свидетельство об упаковывании»
Раздел «Свидетельство о приемке»
Раздел «Движение изделия при эксплуатации»
Раздел «Учет работы изделия»
Раздел «Учет технического обслуживания»
Раздел «Работы при эксплуатации»
Раздел «Сведения об утилизации»
Раздел «Сведения о цене и условиях приобретения изделия».
Данный перечень разделов является общим для всех видов оборудования. Для медицинского оборудования министерством здравоохранения могут быть подготовлены рекомендациями о ведении формуляров. Так, например, Министерство здравоохранения Удмуртской Республики издало Распоряжение от 20 апреля 2016 г. № 434 «О контроле за эффективностью использования медицинского оборудования, находящегося на балансе медицинских организаций УР». В распоряжении четко определены правила ведения формуляров, их состав и требуемая отчетность.
На какое оборудование составляются формуляры?
Как правило, формуляры составляются на:
Перечень оборудования, на которое ведется формуляр, определяет министерство здравоохранения, так, например, в Распоряжении того же министерства от 04 апреля 2017 г. № 506 присутствует четкий перечень оборудования, подлежащего мониторингу эффективности использования, на которое необходимо вести формуляр.
Формуляр на медицинское оборудование является подтверждением качества изделия, соответствия его требованиям нормативной документации, возможности его законного и безопасного использования, а также отражает техническое состояние оборудования в период эксплуатации. Именно его запрашивают сотрудники Росздравнадзора при проверке.
Возможно ли ведение формуляра в электронном виде?
 ГОСТом 2.610-2006 ведение формуляра в электронном виде не запрещается, существует лишь запрет на внесение в распечатанные с электронного носителя формуляры данных от руки. Это означает, что те Лечебно-профилактические учреждения (ЛПУ), кто ведет формуляры в электронном виде имеют право распечатать его, например, в период проверки или запроса проверяющих органов.
ГОСТом 2.610-2006 ведение формуляра в электронном виде не запрещается, существует лишь запрет на внесение в распечатанные с электронного носителя формуляры данных от руки. Это означает, что те Лечебно-профилактические учреждения (ЛПУ), кто ведет формуляры в электронном виде имеют право распечатать его, например, в период проверки или запроса проверяющих органов.
Может создаться впечатление, что заполнение формуляра кроме дополнительных затрат времени ничего не дает, но давайте посмотрим на него внимательнее. Что мы видим: перечень всего оборудования ЛПУ с полными паспортными данными и техническими характеристиками; перечень расходных материалов, необходимых для использования оборудования; комплектующие на изделия; информацию о наработках за периоды эксплуатации и пр. Эта информация может быть очень полезной для принятия таких управленческих решений как:
В связи с масштабными госпрограммами обеспечения учреждений оборудованием и его модернизации, от Минздрава и Росздравнадзора поступает большое число запросов на предоставление различных отчетов об эксплуатации оборудования. Зачастую они разноплановые, имеют много вариативные формы, большой объем исходных данных и короткий срок предоставления. Но как было отмечено ранее все эти данные присутствуют в разных разделах формуляра. Если формуляр ведется в электронном виде и информация хранится структурировано, то решение такой сложной задачи как формирование аналитической отчетности можно возложить на специальную автоматизированную систему учета оборудования (ТОРО).
Автоматизированная система для учета медицинского оборудования. Чем полезна?
Таких программ достаточно много, это специализированные системы управления производственными активами из класса EAM. Мы подробно остановимся на одной из таких систем — это Олимп-ТОРО и расскажем, для чего нужны подобные системы медицинским учреждениям.
Если в вашем ЛПУ имеется лечебное оборудование и его достаточно много, то первое, чем вам может помочь система – это иметь под рукой полный структурированный список всех моделей и марок, которые у вас используются: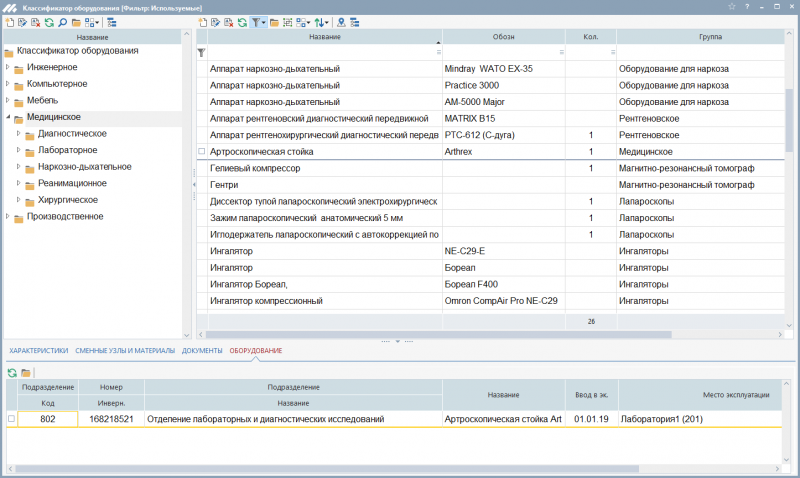
Для каждой марки система покажет перечень оборудования, находящегося в эксплуатации: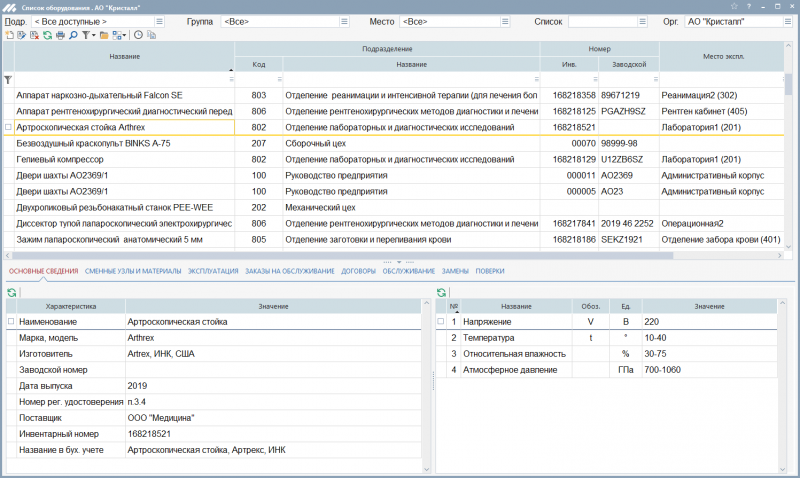
Наличие такого списка позволяет понимать, в каком подразделении находятся изделия, сколько их, какие у них характеристики и в каком они состоянии.
Из представленного списка программа позволяет печатать все необходимые отчетные формы, образующие раздел формуляра «Основные сведения об изделии»: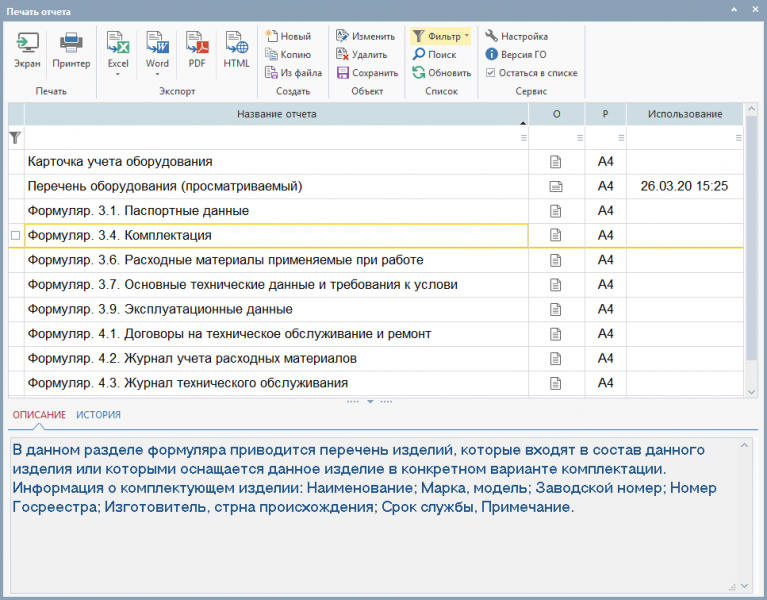
Также вы можете получить из программы и другие разделы формуляра, которые в обязательном порядке должны вестись лечебно-профилактическими учреждениями.
Давайте рассмотрим, как информация вносится в систему, а далее попадает в формуляр:
1. При поступлении оборудования в ЛПУ в системе ответственным специалистом формируется карточка учета куда вносятся все паспортные данные об изделии: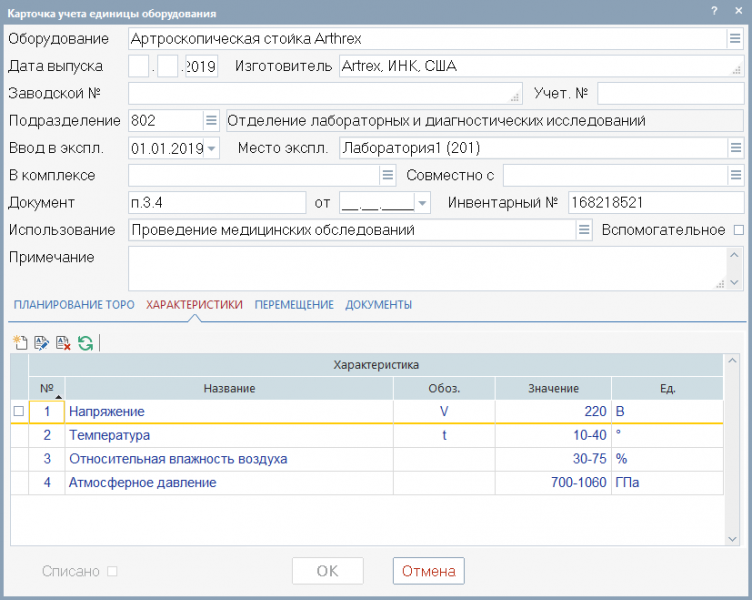
2. Сотрудник подразделения (старшая медсестра) регулярно подает в техотдел отчеты о работе оборудования в которых указывается количество применений, смен работы и объем наработки. Этот отчет вносится в систему, и в результате этого информация о работе оборудования автоматически попадает в печатные формы раздела формуляра «Сведения об эксплуатации изделия», специально вносить эти данные в формуляр уже не нужно. Кроме того, эти отчеты являются основой для автоматического формирования сводного отчета: «Мониторинг эффективности использования оборудования».
3. По аналогичной схеме, создавая в системе Акт выполненного обслуживания, эти данные автоматически попадают в соответствующий раздел формуляра.
Получение формуляра из программы — это всего лишь результат ежедневной работы специалистов, которые в программе делают заявки на обслуживание, формируют акты о техническом состоянии изделий, заключают договора с обслуживающими организациями, фиксируют факты перемещений, наработки, простои, списание оборудования. Другими словами, вся деятельность, связанная с эксплуатацией оборудования, ведется в программе.