качественная реакция на енольную форму

Опыт № 66. Кето-енольная таутомерия ацетоуксусного эфира
Таутомерные формы находятся в состоянии динамического равновесия, одна из таутомерных форм способна самопроизвольно переходить в другую.
При комнатной температуре равновесие устанавливается при содержании 92,5% кетонной формы и 7,5% енольной формы.
Раствор бромной воды
Раствор хлорида железа (III)
2 мл воды приливают
10 капель ацетоуксусного эфира и 1-2 капли хлорида железа (III). Раствор окрашивается с сине-фиолетовый цвет, обусловленный образованием комплексной соли с енольной формой ацетоуксусного эфира.
Затем к раствору прибавляют по каплям бромную воду, раствор обесцвечивается, т.к. бром присоединяется по месту разрыва двойной связи и енольная группировка исчезает. Через некоторое время сине-фиолетовая окраска вновь появляется.
Появление окраски с хлоридом железа (III) объясняется наличием в растворе енольной формы бромзамещенного ацетоуксусного эфира.
Процесс можно изобразить следующей схемой:

Опыт № 67. Реакция на обнаружение углеводов с a-нафтолом (реакция Молиша)
Эта реакция очень чувствительна и позволяет определять даже следовые количества моносахаридов и др. углеводов. Она основана на превращении гексоз при нагревании с серной кислотой в оксиметилфурфурол, пентоз – в фурфурол. Полученные соединения с a-нафтолом дают продукты конденсации окрашенные в красный или синий цвет
Спиртовой раствор a-нафтола
Концентрированная серная кислота
В пробирку наливают
1 мл раствора глюкозы. Далее прибавляют небольшое количество спиртового раствора a-нафтола и не перемешивая содержимое пробирки приливают (осторожно!) концентрированную серную кислоту до образования красно-фиолетового кольца.
Опыт № 68. Доказательство наличия гидроксильных групп в глюкозе
Раствор гидроксида натрия
Раствор сульфата меди
В пробирку наливают
1 мл раствора глюкозы, добавляют несколько капель гидроксида натрия и сульфата меди. Образующийся осадок гидроксида меди (II), при взбалтывании содержимого пробирки, растворяется и получается прозрачный раствор синего цвета.

Растворение гидроксида меди (II) происходит в результате образования комплексного соединения – сахарата меди за счет имеющихся в глюкозе гидроксильных групп.
Чтобы распечатать файл, скачайте его (в формате Word).
Оксокислоты
Способы получения оксокислот
α-оксокислоты получают тремя основными способами:
Гидролиз α,α-дигалогенкарбоновых кислот



ацилхлорид α-оксонитрил α-оксокислота

Присоединение воды к дикетену

Получение γ- и δ-оксокислот

ХИМИЧЕСКИЕ СВОЙСТВА ОКСОКИСЛОТ
Химические свойства α- оксокислот
Для α-оксокислот характерны все реакции карбоновых кислот и карбонильных соединений. Только взаимное влияние карбоксильной и карбонильных групп увеличивает их реакционную способность.
Типичными реакциями α-оксокислот являются:
Взаимодействие с серной кислотой
Реакция декарбоксилирования происходит под действием разбавленной серной кислоты при слабом нагревании с получением альдегидов, а реакция декарбонилирования при нагревании с концентрированной серной кислотой с получением карбоновых кислот:


Глиоксалевая кислота вступает в реакцию Канниццаро:

Химические свойства β- оксокислот
При нагревании все β-оксокислоты распадаются с выделением СО2 и образованием карбонильных соединений:

Химические свойства γ- и δ- оксокислот
Реакции внутримолекулярной циклизации
γ- и δ- оксокислоты обладают всеми свойствами карбоновых кислот и карбонильных соединений, но одновременно для них возможны также реакции внутримолекулярной циклизации:

(этиловый эфир ацетоуксусной кислоты)

Присоединение этилового спирта к дикетену

Сложноэфирная конденсация Кляйзена
Ацетоуксусный эфир образуется при конденсации двух молекул этилацетата под действием этилата натрия:

Ацетоуксусный эфир – классический пример соединения, способного к кето-енольной таутомерии.
Одна из изомерных форм ацетоуксусного эфира содержит кетогруппу (кетоформа), другая – гидроксильную группу у ненасыщенного атома углерода (енольная форма):

Кето-енольная таутомерия относится к прототропной таутомерии, так как она сопровождается переносом протона.
Такое превращение возможно из-за наличия в кетонной форме СН2-группы, расположенной между двумя СО-группами, в результате чего атомы водорода в этой метиленовой группе приобретают значительную подвижность.
Енольная группировка энергетически менее выгодна, чем кетонная. Однако в данном случае при образовании енола возникает:
сопряженная система кратных связей;
внутримолекулярная водородная связь.
Выигрыш энергии за счет этих двух факторов компенсирует в некоторой степени затрату энергии на образование енола, благодаря чему склонность к енолизации возрастает.
В химическом отношении ацетоуксусный эфир способен давать реакции как для кетонной, так и для енольной формы.
Реакции кетонной формы
Восстановление водородом в момент выделения

β- гидроксимасляной кислоты
Присоединение синильной кислоты

Взаимодействие с гидросульфитом натрия

кетоформы ацетоуксусного эфира
При взаимодействии с гидроксиламином образуется оксим кетоформы ацетоуксусного эфира, который является неустойчивым и, теряя молекулу спирта, легко превращается в метилоксазолон:
Енолизуемые карбонильные соединения
Важнейшей особенностью многих карбонильных соединений является возможность обратимого превращения в электронодонорные олефины с высокой реакционной способностью, могущие служить нуклеофилами в огромном количестве важнейших реакций. Именно это свойство делает карбонильные соединения (не все, но большинство) универсальными реагентами в органических реакциях. Получается, что в каждое такое карбонильное соединение встроена кнопка, переключающая электрофил в нуклеофил и обратно. Строго говоря, карбонильные соединения позволяют переключать себя несколькими способами, но именно тот, которому посвящена эта страница основной и самый мощный.
Для того, чтобы эта возможность была, необходимо, чтобы рядом с карбонильной группой был атом углерода, несущий хотя бы один атом водорода. Такой атом углерода может быть один (в альдегидах и многих других карбонильных соединениях) или два (в кетонах). Такие карбонильные соединения имеют две формы – кетонную (всегда так называется, вне зависимости от того, чем реально является карбонильное соединение – альдегидом, кетоном, карбоновой кислотой или ее производным и т.п.) и енольную.

Способность енолизуемых карбонильных соединений к обратимому образованию енолов имеет ряд чрезвычайно важных следствий.
Кето-енольная таутомерия
Кето-енольная таутомерия. Кислотный и основный катализ.
В таутомерном превращении кетона в енол и обратно перемещается только один атом – водород, причем без электронов, то есть протон. Фокус состоит в том, что протон не умеет просто взять – и прыгнуть с одного атома на другой. Вернее, скажем так, умеет, но только в том случае, если эти два атома располагаются настолько рядом, что могут быть связаны водородной связью. Такие случаи в химии бывают, но кето-енольное превращение в простых карбонильных соединерях к ним не относится. Расстояние между углеродом и кислородом довольно значительно, водородной связи между ними нет. Кето и енол – две вполне самостоятельных молекулы, и для того, чтобы превратить одно в другое нужен посредник, который сначала возьмет протон, а затем отдаст его, или наоборот. Такой посредник называется переносчиком протона, и мы отлично знаем, что этот термин описывает хорошо знакомый нам класс веществ – кислоты и основания Бренстеда-Лоури. Переносчик протона, обеспечивающий превращение кетона в енол и обратно (напомню в очередной раз еще один из основных законов химии: прямую и обратную реакцию катализирует один и тот же катализатор по одному и тому же механизму). Посмотрим, как это делают кислоты.
Кислотный катализ таутомеризации

Обратим внимание и на катион в квадратных скобках – интермедиат между енолом и кето-формой. Формально это карбокатион, но реальная структура этой частицы ближе к форме протонированного по кислороду карбонила по очень простой причине – в этой форме все атомы имеют полную электронную оболочку, что всегда выгоднее, чем карбокатион с 6 электронами. Мы уже встречались с этим интермедиатом, например, когда рассматривали кислотный катализ реакции карбонильных соединений с нуклеофилами – протонирование кислорода увеличивает электрофильность карбонильного углерода. В этом одна из сложностей химии карбонильных соединений – одни и те же интермедиаты возникают в разных процессах, и тот же кислотный катализ активирует и присоединение к карбонильной группе, и кето-енольную таутомерию. Одно из последствий этого – кислотно-катализируемая альдольная конденсация.
При некоторой дополнительной дотошности можно усомниться в том, насколько действительно легко протонировать кетон или альдегид, ведь, наверное, это очень слабые основания, и кислота потребуется сильная. Да, это действительно так. Протонировать атом кислорода в карбонильной группе могут только действительно сильные кислоты, например, серная. Но, во-первых, для осуществления катализа нам не нужно, чтобы значительная часть или даже все карбонильное соединение в растворе было протонировано – достаточно только малой части, чтобы кето-енольное превращение стало возможно. Степень протонирования будет определять скорость этого процесса, скорость достижения равновесия. Меньше кислотность реакционной среды – меньше скорость, и все. И все? Нет, не совсем. Даже в присутствии слабых кислот, например, уксусной, равновесие устанавливается, хотя и гораздо медленнее, чем в присутствии сильной кислоты. Дело в том, что протонирование в полном смысле этого слова не обязательно. Достаточно образования водородной связи. Тогда при превращении кето-формы в енол получается что-то типа согласованного перемещения протона при посредничестве слабой кислоты – переносчика протона. Это даже можно изобразить внутримолекулярно, хотя это не обязательно, потому что можно использовать не одну, а две или более молекул переносчика. Обратим внимание на то, что в квадратных скобках изображены именно резонансные структуры, а не равновесие: в резонансных структурах все атомы, включая водороды, остаются на месте, смещается только электронная плотность.

Такой кислотный катализ часто называют общим, потому что он работает всегда вне зависимости от способности кислоты к протонированию, и на него способен гораздо более широкий круг кислот. Скорость взаимопревращения кето-фомы и енола в этом случае зависит от общей концентрации кислоты, так же как и от ее константы кислотности. В химии карбонильных соединений этим видом катализа с удовольствием пользуются, прежде всего потому что этот тип катализа уменьшает вред от побочных реакций, в первую очередь альдольно-кротоновой самоконденсации, в которые часто вступают карбонильные соединения в присутствии сильных кислот.
Основный катализ таутомеризации
Пожалуйста, никогда не говорите “щелочной”, если не хотите прослыть незадачливыми гостями из далекого прошлого, когда серную кислоту называли купоросным маслом. Только оснóвный (не основнóй).
Суть основного катализа кето-енольного превращения – использование основания, которое может превращать как кето-форму, так и енол в сопряженное основание – енолят. Кето-форма – это CH-кислота, а енол – OH-кислота, просто спирт. Сопряженное основание у них одно, мезомерно-стабилизированный енолят, названный так потому что этот анион гораздо ближе соответствует форме с минусом на кислороде, гораздо более электроотрицательном атоме чем углерод. Так как перенос протона обратим, обратим и весь процесс, превращающий кето-форму в енол и обратно.

Какой силы должно быть основание, чтобы вызывать кето-енольное превращение? Здесь та же самая история, как и с кислотным катализом. От силы основания и его концентрации зависит скорость достижения равновесия, но даже более слабые основания по сравнению – по сравнению с чем? С кето-формой, конечно. Но кето-форма это не основание, а кислота. Отлично, именно так, потому что основность мы тоже измеряем константой кислотности сопряженной кислоты, то есть в данном случае как раз кето-формы. Это еще одна причина того, почему в химии принято характеризовать основания Бренстеда-Лоури не константой основности, а константой кислотности сопряженной кислоты. Получается невероятно удобно.
Итак, если мы посмотрим pK простых альдегидов и кетонов, то мы увидим величины порядка 24-26. Имеются в виду значения, полученные в полярном апротонном растворителе, но в реальности реакции альдегидов и кетонов с участием енолятов чаще делают в протонных растворителях типа спиртов или даже водных спиртов. Фокус в том, что мы не знаем надежной оценки pK альдегидов и кетонов в таких растворителях, но можем быть уверены, что это величины, существенно меньшие 24-26. Поэтому для обратимого депротонирования годятся простые основания типа алкоголятов или даже обычных щелочей. Очень часто останавливаются на таком компромиссе, как щелочь (LiOH, NaOH, KOH, Ca(OH)2 и т.п.), но в спиртовой среде (метанол, этанол), а не в водной. Основность гидроксид-аниона в спиртах несколько выше основности этого аниона в воде, а кислотность кето-форм альдегидов и кетонов, наоборот ниже чем в апротонной среде. К сожалению, мы не знаем надежных количественных оценок ни того, ни другого, потому что спирты – очень неприятные растворители для физико-химических измерений, но на качественном уровне понимаем, что разница не так велика, и основности гидроксид-иона в спиртах вполне достаточно для обратимого депротонирования кето-форм и катализа кето-енольной таутомеризации.
Соотношение кетонной и енольной форм
Если мы хорошо понимаем, что такое кислоты и основания Бренстеда-Лоури, а без этого в органической химии вообще никуда не проехать, то вопрос о соотношении кетонной и енольной форм у конкретных соединений поставит нас в тупик своей примитивностью. “Это же очевидно!” – воскликнем мы. Естественно, для того, чтобы ответить на этот вопрос количественно, нам потребовались бы надежные данные, но, как мы знаем, в химии обычно спрашивают не количественную оценку, а качественную, например, просят сравнить то и это, определить качественную тенденцию и т.п.
Если так, то у нас нет проблем. Кетон и енол – кислоты Бренстеда, причем очень забавные, потому что сопряженное основание у них одно и то же – енолят. Тогда все очевидно – протон будет в большей степени принадлежать более слабой кислоте, и чем больше разница в кислотности, тем больше будет разница в содержании кетонной и енольной форм – протон будет сидеть в той форме, кислотность которой меньше.

Получается даже нечто похожее на строгую формулу, ведь константа равновесия таутомерного превращения просто равна отношению констант кислотности енола и кето-формы. Но строгое применение этой формулы наталкивается на труднопреодолимые препятствия, потому что количественные величины этих констант редко известны. Но нам формула и не нужна, а нужно понять, какой формы больше, то есть больше или меньше единицы эта константа.
Не путайте таутомерное равновесие кето-форма – енол и мезомерию енолята.
Как однажды сказал один из деятелей наполеоновской Франции по существенно менее важному поводу: “Это хуже чем преступление. Это ошибка.” Я бы даже сказал, что это не просто ошибка, а мать всех ошибок. Ведь действительно, как похоже:

Но это две принципиально разные вещи. Извиняюсь за напоминание тривиальных вещей, но это действительно очень важно, потому что одна из основных причин ошибок – неряшливое, приблизительное отношение к основным понятиям. Поэтому напоминаю, что кето-форма и енол – две разные молекулы, которые могут превращаться друг в друга с большей или меньшей скоростью в присутствии кислотных или основных катализаторов, и связаны равновесием, которое изображается двойной стрелкой типа “туда-сюда”. Если умудриться получить чистые формы и тщательно исключить любые кислотные или основные примеси (придется, например, отказаться от стекла), а это сделать невероятно трудно, но не невозможно – кето-форма будет сохранять чистоту практически неограниченно, хотя енольная форма не сможет этого сделать, потому что сама обладает заметной бренстедовской кислотностью, а следовательно способна сама катализировать собственное превращение в кето-форму. В равновесных смесях мы можем отлично видеть каждую из форм разными спектроскопическими методами. Каждая из форм обладает своей и очень характерной реакционной способностью.
Енолят с конкретной структурой – одна молекула (ион – это тоже молекула). Структура енолята очень близка к правой граничной структуре, поэтому мы так его и называем, но теория мезомерии обязывает нас изображать и “кетонную” граничную структуру, связав обе обоюдоострой стрелкой, специально зарезервированной для этой теории, и не имеющей никаких других применений в химии. Эта стрелка означает то, что мы не имеем дело с реальными молекулами слева и справа от стрелки, но утверждаем, что реальная структура где-то между этими граничными случаями – не обязательно посредине, возможно даже, что очень близко к одной из них, но все равно для понимания поведения реальной молекулы надо иметь в виду, что каждая из граничных структур несет важную информацию. В случае енолята, например, хотя структура иона енолята очень близка к енолятной форме, но его поведение в реакциях говорит о делокализации электронной плотности. Наилучшим образом это можно было бы увидеть, разглядывая молекулярные орбитали енолята, но как-то у нас это не принято, и придется с этим смириться. Впрочем, немного пониже мы их все-таки порязглядываем.
Существует ли виниловый спирт и другие простые енолы?
Простое правило гласит, что непредельные спирты с гидроксильной группой у двойной связи неустойчивы и самопроизвольно перегруппировываются в карбонильные соединения. Правилу этому очень много лет (оно основано на работах 1877 и 1880 года), в нашей стране оно известно как правило Эльтекова-Эрленмейера, а в остальном мире просто как правило Эрленмейра, и относится оно к большой группе эмпирических правил, унаследованных нами от описательной науки 19 века, когда еще не было никаких представлений о химической связи, электронах и прочих важных вещах, а выводы делались на основе интуитивного обобщения накопленного, ещё не очень обширного экспериментального мтериала. Польза от этих правил для истории органической химии была несомненно, поэтому мы и поминаем их до сих пор, в основном просто чтобы показать, что мы соблюдаем приличия, и не занимаемся каждые 50 лет выбрасыванием с корабля всего, что на нём накопилось. Но правила необходимо переосмыслять на основании и нового материала, и новых понятий и теорий. То же и с правилом Эльтекова-Эрленмейера. Оно не абсолютно, а всего лишь выражает хорошо нам уже известное наблюдение, что для простых енолизуемых карбонильных соединений равновесие енолизации сдвинутов сторону кето-форм. И более того, постольку мы теперь знаем намного больше, то утверждаем, что для превращения виниловых спиртов в карбонильные соединения обязателен посредник, переносчик протона, кислота или основание Бренстеда-Лоури, и что после установления равновесия некоторое количество такого непредельного спирта, или енола, остается и может быть обнаружено с помощью высокочувствительных физико-химических методов, и даже просто по реакционной способности. Но значит ли это, что простые енолы в принципе можно получить как самостоятельные молекулы и даже вещества, и как-то наблюдать за их жизнью. Существуют ли самые простые енолы, например, енол ацетальдегида – виниловый спирт – или енол ацетона – пропен-2-ол?
Да, безусловно существуют, и за последние полвека в литературе описаны многочисленные успешные попытки получения и того, и другого. И вполне доказанным можно считать то, что эти соединения вполне устойчивы, если из системы исключены любые кислоты и основания Бренстеда-Лоури. А вот если они присутствуют, хотя бы в следовых количествах, то простые енолы быстро перегруппировываются в ацетальдегид или ацетон. Получить простые енолы можно многими способами. Самый очевидный – быстрое подкисление енолята в растворе сильной кислотой – в этом случае перенос протона происходит чрезвычайно быстро, и образующийся енол вполне удается увидеть с помощью какой-нибудь быстрой спектроскопии (к сожалению, к таким методам не относится ЯМР – для регистрации спектра ЯМР по принципиальным причинам требуется значительное время, скажем очень приблизительно, что это время измеряется секундами, в то время как таутомерное превращение простых енолов в среде, где есть переносчики протонов, а без них подкислить ничего нельзя, – измеряется долями миллионных долей секунды). Но есть много очень шустрых спектроскопических методов, и с их помощью виниловый и пропениловый спирты были не только обнаружены, но и измерены кинетические параметры перегруппировки. Метод оказался хорош, и если бы удалось добавить настолько точное количество кислоты, чтобы каждая молекула енолята превратилась бы в енол – то можно было бы попробовать получить чистый енол в отсутствие катализаторов. Увы, это за пределами возможностей эксперимента – ничтожный избыток кислоты создаст кислотный катализ, а ничтожный недостаток – основный, ведь непрореагировавший енолят – очень сильное основание, и будет сам катализировать превращение енола в кето-форму.
Исключить сильные кислоты и основания удавалось более инструментальными методами, например, вакуумным пиролизом (очень быстрым нагреванием разреженного пара) этиленгликоля – при этом происходит термическое элиминирование воды. В такой системе в парах при пониженном давлении виниловый спирт устойчив минутами и даже часами. Но и там есть вода, и хотя в парах вероятность встречи молекулы винилового спирта и молекулы воды невелика, а вода это слабая кислота Бренстеда-Лоури, и вполне способна вызывать более медленное, но неотвратимое превращение енола в кето-форму. Сконденсировать эти пары и получить жидкий виниловый спирт тоже идея спорная, потому что сам виниловый спирт является слабой кислотой, более слабой чем вода, но все равно способной катализировать превращение других молекул винилового спирта в ацетальдегид.
Но в высоком вакууме, где молекул мало и вероятность их встречи пренебрежимо мала, виниловый спирт и другие простые енолы, скорее всего, как говорят, бесконечно устойчивы – то есть не превращаются с измеримой скоростью в свои таутомеры. Одно из надежных доказательств этого найдено ни много ни мало – в космосе. Еще в 2001 году было обнаружено, что в нашей родной галактике в межзвездном пространстве полно винилового спирта, Полно, естественно, в космическом смысле – от одной молекулы до другой пришлось бы пару сотен лет пешком идти. Вот и хорошо, но молекулы-то есть, существуют там тоже в космической шкале времени, возможно, найдутся и такие, которые динозавров наших из своего далека видели (не придирайтесь к скорости света, у нас не очень большая галактика, плюс-минус 50-100 тысяч лет это не принципиально). И не превращаются в ацетальдегид (давно бы превратились, если бы это происходило). Обязательно кто-нибудь заметит – да ведь там чертовски холодно, мало ли какая молекула может выжить при температуре жидкого гелия. Верно, но здесь весь вопрос в том, сколько выжить. Таутомеризация – это просто перемещение протона, и если бы этот процесс мог происходить без посредников, то низкая температура ему бы не помешала.
Реакции карбонильных соединений, происходящие за счет енола
Содержание енолов в простых альдегидах и кетонах – величины порядка от одной тысячной до одной десятимиллионной части процента. И что – мы будем обсуждать такую безделицу?! Какой смысл обсуждать такие ничтожные примеси? Ведь если мы возьмем ацетальдегид, ацетон или циклогексанон, даже очень чистый, из хорошей реактивной лавки типа Мерк-Сигма-Олдрич, обслуживающей весь мир первосортными реактивами, то будем на банках иметь чистоту 97-98%. А что все остальное? Вода, и еще много всякой дряни и ерунды. И такой чистоты обычно хватает для большинства работ. Если раскошелимся, можем купить и высокочистые реактивы – 99%, даже 99,9%, – и все равно 0.1% примесей. А енола – в сто, тысяча, и даже больше раз меньше чем этих никому не интересных примесей. Почему нас не волнуют эти анонимные примеси, но волнует нечто в тысячи раз более ничтожное?
Прежде всего потому что у енола есть два свойства, которые делают ничтожную примесь критически важной.
Посмотрим поподробнее на некоторые важные реакции, в которых енолизуемое карбонильное соединение реагирует через енол.
Рацемизация
Если у нас есть альдегид или кетон (или любое другое карбонильное соединение), которое имеет рядом с карбонильной группой асимметрический атом углерода и хотя бы один водород на нем, и имеем это соединение в виде одного из энантиомеров, то нам придется соблюдать очень строгие меры предосторожности, иначе мы будем терять оптическую чистоту, по ка не получим рацемическую смесь.
Происходит это по очевидной причине – при образовании енола асимметрический атом углерода становится sp2-гибридным, плоским, и обратное присоединение протона при превращении енола в кето-форму происходит с равной вероятностью снизу и сверху этой плоскости. В результате, какой бы энантиомер мы не взяли, в конце концов они превратятся в рацемат. Поэтому этот процесс называется рацемизацией.

Для рацемизации нужен кислотный или основный катализ, который может прийти, откуда не ждали, например, от стенок стеклянной банки (стекло – силикат натрия, обладает весьма приличной основностью), или от силикагеля (силикагель – весьма приличная кислота в основном по причине наличия на поверхности гидроксильных, так называемых силанольных групп) при попытке очистить соединение хроматографией. Поэтому и получить и сохранять оптически активные альдегиды и кетоны очень непросто. Но не невозможно, что в успехом демонстрирует нам природа, так как, например, все углеводы, включая глюкозу и фруктозу (но не сахарозу), являются энантиомерно чистыми альдегидами и кетонами и один из стереогенных центров в этих соединениях находится на атоме углерода, участвующем в енолизации. Почему это так, мы разберемся, когда дойдем до углеводов.
Еще один пример оптически чистых карбонильных соединений, имеющих енолизуемый стереогенный центр – природные аминокислоты, из которых состоят белки и пептиды. Это не альдегиды и кетоны, поэтому сильно вперед забегать не будем, просто отметим, что в химии аминокислот тщательно избегают реакций, в условия которых входит действие сильных оснований в протонодонорной среде. Именно потому. что как огня боятся рацемизации, катализируемой основанием. Почему в этом случае не боятся кислотно-катализируемой рацемизации, тоже разберемся в своем месте. А для простых альдегидов и кетонов и кислоты, и основания одинаково опасны.
Дейтерообмен
Другое интересное следствие обратимого образования енола – дейтерообмен α-протонов. Он происходит в том случае, если карбонильное соединение растворить в протонодонорной среде, в которой обменивающиеся протоны заменены на дейтерий, и в присутствии кислоты или основания. Обмениваются только протоны на соседних с карбонилом атомах углерода, например:

Обратите на это внимание – протоны (дейтероны) идут не от кислоты или основания, а от среды. Кислота или основание – катализаторы, их может быть очень мало, и они могут быть даже самые обычные, хотя если есть под рукой дейтерированные, то и хорошо – это чуть-чуть ускорит дело, а почему, скоро разберемся. Средой обычно служит тяжелая вода или ее смесь с апротонным растворителем (только не берите ацетон. ). Но можно, в принципе, взять спирты с дейтерированным гидроксилом (остальные протоны вполне могут быть обычными), или уксусная кислота или что-то подобное. См дейтерообмен идет по очевидной причине, хотя для кислотных и основных условий придется написать немного разные схемы. В кислотном катализе енол будет протонироваться кислотой, переносящей протон (дейтерон) от протонодонорной среды, например, воды. В основных условиях, протонироваться будет уже енолят в равновесии сопряженной кислотой основания, опять таки, переносящей протон (дейтерон) от среды.

А теперь внимание! Это очень жесткий тест на то, понимаем мы или нет, что такое равновесие. Равновесие – это одновременно и страшная, почти непреодолимая сила, и одновременно чрезвычайно грубый инструмент Природы. Для работы с равновесием Природа выдала нам единственный инструмент – принцип Ле Шателье, и другого нет и не предвидится. Это обстоятельство просто бесит, но придется смириться.
Так вот, если у нас в кетоне или альдегиде несколько α-протонов (в ацетальдегиде и ацетофеноне три, в циклогексане 4, в ацетоне аж 6, и больше не бывает), то мы ни при каких обстоятельствах и ни в каких обратимых условиях не сможем обменять только часть из них, например, один. Не поможет ни недостаток или избыток чего бы то ни было, ни температура, ни время реакции, ни заклинания, ни проклятия, ни помощь потусторонних сил, ничего. Скорости и константы равновесия для недейтерированных и дейтерированных субстратов отличаются, но не сильно и этим можно пренебречь, считая вероятность обмена просто равной вероятности встретить молекулы с разным числом атомов дейтерия, а это просто их соотношение в растворе. Например, если у нас обмениваемых протонов два, а мы хотим один и возьмем один эквивалент источника дейтерия (полмоля тяжелой воды на моль кетона) то в самом начале процесса у нас будет обмениваться один протон, но как только монодейтерированный продукт накопится хотя бы в количестве 10%, вероятность обмена второго протона станет вполне существенной, округленно 0.1 и начнет накапливаться дидейтерированный продукт, и далее все пойдет со все более сближающимися скоростями (или вероятностями, что в данном случае одно и то же).
Единственно, что мы можем сделать, это обменять все α-протоны на дейтерий. И это не совсем просто, если мы хотим достигнуть хорошей степени обмена. Сразу поймем, что 100%-ного обмена в одной реакционной смеси достичь невозможно, так как протоны, выходящие из объекта обмена включаются в общее равновесие, и не дают ему прийти к полной замене протона на дейтерон. Скажем, если у нас два протона и мы взяли 100-кратный избыток воды (то есть соотношение D:H = 100:1), то обменять больше 99% у нас не получится. И это отличный результат – редко нужно больше. Но если нужно 99.9, то нам потребуется 1000-кратный избыток тяжелой воды. И хотя тяжелая вода не самый дорогой на свете реактив, но даже 100-кратный избыток может остановить многих, а 1000-кратный. Такой несуразный избыток, скорее всего, остановил бы всех желающих даже в некоторых странах Персидского залива, где деньги, выделяемые на науку, измеряются порядками числа Авогадро, и уже точно ни в какие ворота не лезет в странах, где соответствующие фонды имеют порядок постоянной Планка.
Средство от этого хорошо известно вообще в любых областях, где имеют дело с равновесием. В экстракции, хроматографии, ректификации, делении изотопов и т.д., и т.п. всегда делают одно и то же – то, что не удается достичь одним равновесием, получается, если процесс достижения равновесия последовательно повторить несколько, иногда много раз. Так и здесь. Первый раз берем 10-кратный избыток тяжелой воды. Получим что-то около 10% остаточных протонов, или дейтериевую чистоту в 85-90%. Выделяем из реакционной смеси кетон с таким содержанием, и повторно запускаем с тем же количеством тяжелой воды. В этот раз по отношению к остаточным протонам избыток источника дейтерия получится уже почти 100-кратным, соотвественно и результатом будет остаточное содержание протонов сильно ниже 1%. Выделяем, пускаем третий раз в тех же условиях. По отношению у остатоным протонам избыток дейтерия станет уже ближе в 1000-кратному, результат будет соотвествующий, и это всего за три раза с суммарным избытком всего 30-кратным. Более того, мы сможем еще сэкономить, если будем повторять процесс с новыми порциями кетона, тогда оставшуюся тяжелую воду с 3 стадии, содержащую небольшое количество обычных протонов, используем на второй в следующий раз, а со второй – на первой. Вот как-то так приблизительно и делают.
Зачем? Например, для того чтобы делать растворители для ЯМР. Все современные приборы (современные = последние лет 40) требуют использования дейтерированных растворителей не только для того, чтобы протоны растворителя не мешались, но и для автоматической настройки параметров прибора. Все известные растворители имеют дейтерированные версии, которые можно купить. Но стоят они все очень по-разному. Есть более доступные (дейтерохлороформ, гексадейтероацетон, гексадейтеродиметилсульфоксид, тяжелая вода), а есть очень дорогие (дейтерированные ТГФ, дихлорметан, диметилформамид и т.п.). Разница в цене определяется очень просто – все, что можно получить прямым дейтерообменом, приблизительно так, как мы это только что разобрали, стоят, условно говоря, по цене дейтерия, потому что сам процесс довольно прост и эффективен, а первичный источник дейтерия, тяжелая вода, производится атомной промышленностью в неограниченных количествах. А те, что нельзя получить прямым дейтерообменом, а требуется многостадийный синтез с использованием меченых реагентов, дороги, потому что приходится оплачивать сложную препаративную работу.
И когда в следующий раз будете делать образец для ЯМР, растворяя свое вещество в дейтерохлороформе или дейтероацетоне, обратите внимание на этикетку растворителя, где всегда указаны остаточные протоны. И обратите внимание на то, что 100%-но чистые дейтерированные растворители почти никогда не используются, хотя есть в продаже, но очень дороги потому что прямым дейтерообменом можно получить какую угодно чистоту, хоть 99,99999%, но потребуется на это очень много повторов равновесия обмена.
Бромирование

Теперь представим себе, что мы прибавляем бром к чистому кетону или альдегиду, или к раствору в каком-нибудь чистом апротонном растворителе. Тогда будет любопытный эффект – моментально прореагирует только тот енол, который содержится в карбонильном соединении согласно равновесию. И все – реакция остановится: енола больше нет, а просто кетон или альдегид в кето-форме с галогеном не реагирует – это электрофил, а не нуклеофил, а электрофил с электрофилом не реагирует.
Очевидно, что это сильно завышенные значения. Если учесть реальное содержание енола в этих кетонах, то завышение в тысячи и десятки тысяч раз. Мышь перепутали со слоном. Даже не мышь, а таракана.

Теперь попробуем понять, как нужно делать реакцию бромирования енолизуемого кетона или альдегида, чтобы получить продукт бромирования. Сразу замечу, что в альдегидами это не так чисто и удобно, как с кетонами, из-за побочных реакций, в частности из-за окисления альдегидной группы, но в первом приближении можно не обращать на это внимание. Возьмем, например, ацетон, и попробуем его пробромировать, добавив эквивалент брома. Увидим любопытный эффект. Сначала получится раствор брома в ацетоне и он довольно долго будет стоять безо всяких изменений. И в какой-то момент вдруг моментально вскипит и обесцветится.

Такая постановка эксперимента дает интересный и поучительный эффект, но когда цель эксперимента не позабавиться, а получить конкретный продукт с хорошим выходом, она не годится – нормальные химики не любят реакций, которые сначала не идут, а потом вылетают в окно вместе с плачущим экпериментатором (плачущим – потому что α-бромкетоны почти всегда являются слезоточивыми веществами). Но тот же механизм отлично подсказывает нам, как сделать так, чтобы никто никуда не вылетел, а реакция шла “быстро и с высоким выходом” (это популярнейший мем органического синтеза). Нужно обеспечить постоянную и приличную скорость кето-енольного превращения, сразу взяв подходящий катализатор в более-менее постоянной концентрации. И тогда нам останется просто прикапывать бром с такой скоростью, чтобы он расходовался. Прекрасно, таким катализатором может быть, например, достаточно слабая уксусная кислота, которую можно еще и взять в качестве растворителя – тогда и концентрация будет постоянной. Так и делают – посмотрите на странице про методы в соответствующей вкладке.
Галоформная реакция
Если бромирование идет через енол, то как идет галоформная реакция – ведь это почти то же самое. Енолизуемый кетон, правда не любой, а только метилкетон в присутствии галогена (хлора, брома, иода), взятого в избытке, но минимум в трехкратном количестве, и обязательно в присутствии водной щелочи, дает необычные продукты, расщепляясь в галоформ и соль кислоты на 1 углерод короче. При этом, нужно различать препаративную галоформную реакцию, когда мы ее делаем для того чтобы превратить кетон в кислоту (см. на странице методы), и качественную галоформную реакцию, которую в старину, когда реакции ставили при свечах и под пение псалмов, использовали как пробу на наличие некоторых веществ в моче вьючных верблюдов. Качественную галоформную реакцию дают не только метилкетоны и ацетальдегид, но и соединения, которые могут окисляться в метилкетоны или ацетальдегид галогеном в щелочной среде, например, спирты типа этилового и изопропилового. После изобретения всяких спектроскопий эта реакция даром никому стала не нужна, а препаративно ее использовать нельзя, потому что щелочной галоген, на самом деле, очень плохой окислитель, слабый и неселективный. Для качественного теста это не важно – окислится 5%, выпадет галоформ, – тест положительный, зачет. Но для препаративных целей выход в 5% с параллельной возможностью этим галогеном еще куда-нибудь заехать совершенно неприемлем. Поэтому лучше забыть про качественную галоформную реакцию, как про предание старины глубокой.
А вот препаративная галоформная реакция – вполне полезная реакция, у которой нет хорошей (то есть простой, дешевой и эффективной) замены даже в самой наисовременной химии. В отличие от бромирования, которое идет в условиях кислотного катализа или автокатализа, галоформная реакция идет в условиях основного катализа. Так, но мы недавно разобрались в том, что катализ не влияет на равновесие, в том числе на соотношение енолов, если их два, как в несимметричных кетонах, енолизуемых в обе стороны. И как же тогда получается, что при бромировании получается более замещенный бромкетон, а в галоформной реакции – продукт из менее замещенных бромкетонов.

Стоит сказать, что хотя мы пишем чистое галогенирование по метилу даже в несимметричных кетонах, у которых с другой стороны есть CH2-группа, реально такой селективности достигнуть трудно. Поэтому галоформную реакцию гораздо чаще применяют к метилкетонам, у которых нет такого соблазна, и с другой стороны или ароматическое кольцо, или двойная связь, или что-то третичное, неенолизуемое. Гидроксид все же очень маленький и недостаточно сильный как основание, чтобы давать чистое депротонирование по менее замещенной стороне, то есть результат, который мы получаем (и то не на 100%!) при генерации енолята действием очень сильного и громоздкого LDA.
Последняя стадия галоформной реакции довольно интересна, потому что мы в ней встречаем старого знакомого в новой роли, и убеждаемся, что поведение реакционноспособных частиц очень сильно зависит от условий, в которых они находятся. После образования тригалогенкетона карбонильная группа в этом соединении становится чрезвычайно электрофильной – тригалометильная группа является сильным индективным акцептором. Поэтому это соединение быстро присоединяет нуклеофил, гидроксид-ион. Обратимо образуется тетраэдрический интермедиат, у которого, казалось бы, нет никаких возможностей во что-то превратиться дальше. Но такая возможность есть, и это отщепление тригалометильного карбаниона, неплохо стабилизированного индуктивным эффектом трех галогенов, что делает его достаточно приличной уходящей группой. В SN2-химии мы не видели такой уходящей группы (на самом деле и там такое бывает, но это очень большая редкость), но в химии карбонила закономерности ухода от тетраэдрического интермедиата не такие жесткие, как в SN2, и уйти может почти любой анион, который хоть немного делокализован и стабилизирован. Мы еще увидим огромное множество таких примеров. Но главная проблема не в этом, а в том, что мы уже однажды видели трихлометильный анион, и узнали про него, что он быстро и необратимо распадается на дихлоркарбен и хлорид – вспомните реакцию алкенов с хлороформом и крепкой щелочью в межфазных условиях, которая приводит к образованию дихлорциклопропанов. Почему же здесь такого не происходит? Да просто потому, что тригалометильный анион в галоформной реакции выходит в водную среду с невысокой основностью (в галоформной реакции не используют больших избытков щелочи) и немедленно протонируется, ведь это все же очень сильное основание, карбанион, стабилизированный всего-навсего индуктивными акцепторами. Образование сопряженной кислоты тригалометильного аниона, то есть галоформа HCX3 происходит в этих условиях быстрее, чем α-элиминирование хлорида. А галоформ, как только образуется уходит в отдельную фазу, или жидкую (хлороформ или бромоформ), или кристаллическую (иодоформ) и покидает поле битвы.

Тут стоит заметить еще одну занятную вещь. Иногда подмечают, что после ухода тригалометильного аниона из тетраэдрического интермедиата формально образуется карбоновая кислота, куда более сильная кислота чем вода, и, дескать, именно она протонирует тригалометильный анион по праву более сильной кислоты. Это замечание столь же глубокомысленно, сколь и бессмысленно, потому что культурное обращение с кислотностью и основностью по Бренстеду-Лоури требует признания, что источником протона в протонодонорной среде, например, в воде, всегда является сама среда и ее основные компоненты – сопряженные кислоты и основания (в воде это вода и гидроксид, или вода и гидроксоний), а прямой передачей протона можно пренебречь. Другое дело, если бы среда была апротонной, и там нет альтернативы прямому переносу протона от кислоты к основанию, но тогда и реакция пошла бы не так. Это замечание выглядит довольно неприличным занудством, но уверяю вас, вы много приобрете, если научитесь корректно работать с кислотными и основными протонодонорными средами. Игра стоит свеч.
Нитрозирование
Понять, что делает реакция нитрозирования енолизуемых карбонильных соединений в нашем, в общем-то вводном курсе органической химии, довольно трудно. Это очень частная реакция, не имеющая широкого применения. Если нам повезет, мы возможно встретимся с этой реакцией во 2 семестре, если решим разобрать исторически очень важный, но в наше время малополезный метод синтеза пирролов по Кнорру, где эта реакция используется как источник α-аминокетонов.
В этой реакции используют только енолизуемые кетоны. Альдегиды легко окисляются в условиях нитрозирования, потому что нитрозо-соединения – это окислители, работающие по механизму гидридного переноса, а это один из основных механизмов окисления альдегидной группы (второй – свободнорадикальный).
Нитрозированием вообще называется введение в органические молекулы нитрозо-группы NO. Это очень странная группа с очень необычной химией, и мы коснемся ее только мимоходом. Источником нитрозо-группы в органических молекулах обычно является электрофильное нитрозирование – реакция органических нуклеофилов с нитрозоний-катионом NO + или ковалентными нитрозо-соединениями с уходящими группами типа нитрозилхлорида NOCl, нитрозилбромида NOBr, азотистого ангидрида ON-O-NO и др. Все эти молекулы элементарно образуются при действии на нитриты сильных кислот – анион кислоты становится уходящей группой в нитрозосоединении, а если анион не имеет заметной нуклеофильности, как например, анион серной кислоты, то получается азотистый ангидрид.
Если в этот момент рядом находятся енолизуемые кетоны, то любая из этих молекул легко реагирует как электрофил. Все эти частицы и даже сам нитрозоний-катион – довольно слабые электрофилы, например, они не реагируют с большинством ароматических соединений кроме самых реакционноспособных типа фенолов и анилинов. Во 2 семестре мы еще посмотрим, что получается там с нитрозированием. В любом случае, это неплохая аналогия: фенолы и енолы это очень близкие по структуре и реакционной способности соединения.

Образующийся нитрозокетон – очень интересная штука, потому что он очень похож на 1,3-дикетоны, которыми мы еще займемся, и точно также должен быть склонен к таутомерии, причем можно нарисовать два разных таутомера. Сразу заметим, что оставаться в нитрозо-кето-форме, так как нарисовано на схеме реакции, у него нет никаких шансов. Нитрозо-группа – явно очень сильный акцептор, и индуктивный, и мезомерный. Она очень похожа на карбонильную, но имеет в составе более электроотрицательный азот вместо углерода. Поэтому атом водорода на углероде между двух сильных акцепторов обречен иметь немаленькую кислотность, pK такой CH-кислоты не может быть выше 10, а скорее всего и существенно ниже. Поэтому в равновесии такой формы не может быть много (посмотрите еще раз обсуждение соотношения таутомеров на вкладке выше). Но перебраться он может как на кислород нитрозо-группы (нитрозо-оксимная таутомерия), так и на кислород карбонила (обычная кето-енольная таутомерия). Выбрать между этими двумя формами непросто, и на основании общих соображений не получится. Один фактор учесть можно – водородную связь внутри молекулы с образованием шестичленного цикла – это всегда очень выгодно, и ложится увесистой гирькой на чашку весов имеющего такую особенность таутомера. В оксимной форме образование такой связи очевидно и легко, в альтернативной нитрозоенольной форме могут быть еще и два диастереомера – (E) и (Z), и водородную связь может иметь только один из них, следовательно это уж точно не проще чем в оксимной форме. Так или иначе именно оксимная форма преобладает.

Результат интересный – получается, что нитрозирование енолизуемых кетонов дает моно-оксим дикетона. В принципе, его можно было бы гидролизовать в 1,2-дикетон, но есть более надежный способ синтеза этих соединений. С другой стороны, можно добавить гидроксиламин и получить полный оксим дикетона, а это очень интересные соединения. Из метилэтилкетона при этом получается знаменитое вещество – диоксим диацетила, хорошо известный под названием диметилглиоксим (гли – от глиоксаля, так исторически называется простейший диальдегид, этандиаль) он же реактив Чугаева. 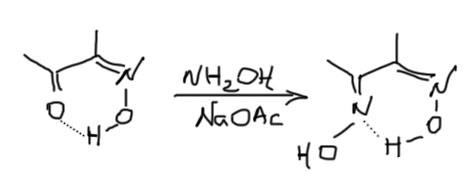
Реактив Чугаева – совершенно потрясающе интересный хелатный лиганд. Особенно хорошо известен его комплекс с никелем, имеющий кроваво- красный цвет. Цвет хорош, но еще красивее структура комплекса, который образован двумя моноанионами лиганда, образующими координационную сферу металла таким образом, что один лиганд дополнительно сцеплен с другим за счет очень прочных водородных связей, и такая структурра образуется из-за очень точной пространственной подгонки обоих лигандов друг под друга, или, как это часто называют, самокомплиментарности. Хотя водородные связи обычно считают межмолекулярными, но здесь они реализованы в одной молекуле комплекса, фактически связывая два отдельных лиганда в макроциклический общий лиганд.

Реакция с диоксидом селена
Окисление диоксидом селена носит название реакции Райли (Riley) в честь первооткрывателя. Довольно полезная реакция, но не очень популярная, потому что диоксид селена редко можно найти в органической лаборатории, и нужно заранее озаботиться покупкой этого реактива, а это довольно серьезный аргумент – обычно, когда нужно что-то сделать, ищут реактивы у себя на полках и спрашивают у соседей. Ничего не найдя, бросают замысел, или выбирают другую дорогу. К тому же с селеном связано много подсознательных страхов – считается, что соединения селена имеют отвратительный запах и очень токсичны. Некоторая правда в этом есть, хотя это не относится к оксидам селена и их производным, да и сам селен – вполне безобидное вещество.
Диоксид селена и селенистая кислота – аналоги сернистого ангидрида и сернистой кислоты, но свойства и реакционная способность у этих близких родственников драматически различаются. Производные черырехвалентной серы – скорее нуклеофилы, а сам сернистый ангидрид – удивительно малореакционноспособное вещество, из-за чего его часто применяют в жидком виде как растворитель, который сам никому и ничему не мешает. А производные четырехвалентного селена обладают весьма богатой и разнообразной реакционной способностью, в первую очередь это вполне неплохие электрофилы, но еще они умеют играть в сложные игры согласованных реакций, участвуя в циклических переходных состояниях с одновременным перемещанием электронов и связей. Диоксид селена – совершенно уникальный окислитель, осуществляющий превращения, которые другими способами потребовали бы многостадийных цепочек. У диоксида селена есть два любимых реакционных центра – сильнодонорные двойные связи и аллильные атомы водорода, – которые он ловко выцепляет из очень сложных структур, и не обращая внимание ни на что другое, превращает в очень полезные продукты. Такие свойства очень нравятся синтетикам. Окислители вообще обычно довольно грубые и бесцеремонные ребята, и каждый раз в тех случаях когда нужно что-то селективно и аккуратно окислить в сложной молекуле, всегда приходится основательно напрягаться, чтобы не окислить что-то лишнее или не переокислить то, что нужно, потому что почти любой окислитель втайне или откровенно мечтает окислить любое органическое вещество до угольного ангидрида и воды.
Диокид селена же не таков – механизмы его действия очень специфичны и аккуратны, они развиваются в основном не как окислительные (а окисление – это обычно банальный грабеж, состоящий в том, чтобы оторвать и присвоить электроны или атомы какие-нибудь вместе с их электронами), а как обычные реакции замещения, присоединения, согласованной еновой реакции, и изменение степени окисления наступает как формальный результат образования и разрыва связей.
Кроме того, это еще и один из первых настоящих реагентов в органическом синтезе в современном смысле этого понятия. Реакция была открыта аж в 1932 году, то есть в то странное время, когда органики обнаружили, что они занимаются этим делом уже приблизительно сто лет, и за это время ничего не поняли, хотя уже перепробовали все доступные им вещества и открыли все возможные реакции между ними, и им остро нужно, во-первых, наконец перестать просто смешивать все подряд и задуматься о том, как эти реакции идут (из чего тут же родилась теория органической химии, то есть наука о механизмах), а во-вторых, выйти из органических лабораторий и зайти к соседям неорганикам в поисках новых реактивов. О, это был непростой шаг. До этого (а многие верят в это до сих пор) органики были совершенно уверены в том, что их химия совершенно особенная, главная, единственная, заслуживающая названия “химия”. Обратите внимание на тот забавный факт, что у неорганической химии вообще нет названия – это просто не органическая химия, все остальное; то есть есть на свете только одна настоящая химия со своим собственным названием. Это удивительно, это все равно, как если бы, например, в языке не было бы слов для каких-то вполне важных вещей, например, вместо “мужчина” говорили бы “неженщина”, а вместо взрослый человек – “неребенок”… И ведь и смысл какой-то в этом можно было бы легко найти (возможно, даже есть такие языки на свете, где это именно так, или как-то похоже, но я в этом не уверен). Но вот с неорганической химией так и получилось, и никто не против. Они, коварные неорганики, просто догадывались, что разок попробовав что-то откровенно неорганическое, органики увлекутся и сами того не заметив, погрузятся в совершенно неисчерпаемые запасы неорганических соединений, и что из этих робких опытов вырастут современный синтез и катализ, и химия координационных соединений, и химия материалов, и что все это ускоренными темпами начнет вытеснять классическую органическую химию на обочину прогресса. В современных журналах, как бы они ни назывались, вы уже с изрядным трудом сможете понять, о какой химии там говорят, но точно не об органической. Это поучительная история, которая показывает, что пафосное название не всегда гарантирует блестящее будущее.
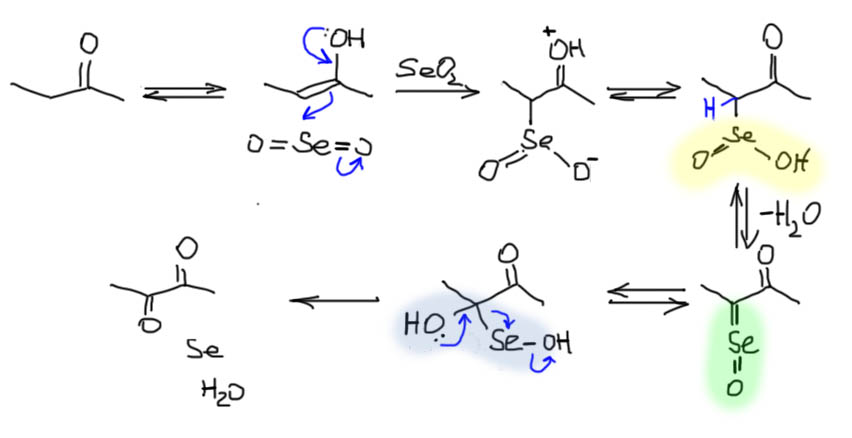
Вернемся к окислению енолизуемых карбонильных соединений диоксидом селена. За уже почти сто скоро как сто лет никто не удосужился изучить механизм этой реакции, но это не помешало целым двум мощным нобелевским лауреатам, Элайасу Кори и Барри Шарплесу сцепиться по поводу этого механизма. С тех пор в литературе попеременно встречается то один, то другой механизм, без попыток выяснить, который из них правильный. Мне более разумным кажется механизм Шарплеса, к тому же он хоть как-то обоснован. Он довольно прост, хотя и содержит пару не очень внятных стадий. Но начинается все просто реакцией электрофильного диоксида селена с равновесным енолом, и, как всегда в таких случаях, если кетон несимметричный, то преобладает более замещенный енол. Отдельный вопрос заключается в том, как обеспечивается катализ кето-енольного превращения в реакции диоксида селена, которую часто проводят в инертном растворителе типа диоксана. Ответ, видимо, простой – сам диоксида селена, видимо, всегда содержит примесь селенистой кислоты, обладающей вполне достаточной кислотностью для того чтобы обеспечивать приемлемую скорость кето-енольного превращения. Итак, присоединяем диоксид селена к двойной связи енола и получаем, после обычного перемещения протона, некий интермедиат, у которого рядом с карбонилом остаток селенистой кислоты (это называется кетоселениновая кислота). Тут мы замечаем уже знакомый нам атом водорода с повышенной кислотностью между двумя акцепторами, и видим неплохую перспективу ухода этого протона с образованием нуклеофильного центра. Но вот проблема – это-то нам как раз и не нужно – нам нужно окислять этот атом, а не восстанавливать, а минус на атоме – признак восстановления. Как быть? Спасение приходит откуда не ждали, потому что дальше происходит нечто, что иногда называют перегруппировкой Пуммерера (до собственно перегруппировки дело не доходит, но некоторая аналогия, заключающаяся в неожиданном изменении реакционной способности атома углерода с обычной нуклеофильной енольно-енолятного типа, на электрофильную из-за того, что между этим атомом и селеном возникает нечто похожее на двойную связь за счет ухода от селена уходящей группы (в этом случае воды). Вот это ровно то, что нам нужно, так как атом углерода в одночасье сделавшийся из нуклеофила электрофилом, спокойно присоединяет воду, а селеновый остаток, который незаметно для себя уже восстановился от Se(4+) до Se(2+), немедленно разваливается на металлический селен и воду. Схема не лишена признаков легкого мошенничества, но таковы многие органические механизмы, и пока никто не возражает, сойдет.
И еще много чего, например, кислотно-катализируемая альдольная конденсация и т.п.
И немало других реакций карбонильных соединений зависят от того, что в енолизуемых карбонильных соединениях содержится небольшое количество енола. Этот енол, выражаясь немного посовременнее, обслуживает важный канал превращения карбонильного соединения в продукты разнообразныз реакций, в основном через электрофильное присоединение к енолу.
Упомянем только еще одну реакцию – кислотно-катализируемую альдольную конденсацию. Почему только кислотно-катализируемую? Потому что более распространенная альдольная конденсация, катализируемая основанием, идет не через енол, а через енолят, а это немного другая история, которой мы займемся отдельно.
В кислотно катализируемой альдольной конденсации нуклеофилом (метиленовой компонентой) является равновесный енол, но что является электрофилом. Иногда, увы слишком часто, говорят, что само карбонильное соединение. Это безусловно не так, потому что, если бы это было так, енолизуемые карбонильные соединения превращались бы в альдоли просто при стоянии. Но ничено подобного не происходит – и ацетальдегид и ацетон не превращаются в свои альдоли. Ацетон вообще стоит годами в закрытой таре без видимых изменений, а ацетальдегид медленно полимеризуется, но не в альдоль и не через альдоль.
Придется вспомнить, что карбонильная группа тоже изменяет реакционную способность в условиях кислотного катализа – увеличивает электрофильность или за счет протонирования по кислороду (сильный эффект, требует присутствия сильных кислот), или за счет образования водородной связи по атому кислорода (слабый эффект, для этого достаточно слабых кислот, желательно в избытке, но в альдольной конденсации этот тип кислотного катализа почти не работает). Возможен еще катализ не протонными кислотами, а кислотами Льюиса типа безводного хлорида цинка, но мы не будем трогать этот путь, так как там очень трудно разобраться в каше возможных путей и продуктов. Просто пока заметим, что этот путь является основным, если в качестве метиленовой компоненты берут не енол (еще раз повторю – енол взять нельзя, можно только смиренно дождаться милостей от никак не зависящего от вас равновесия), а стабильные и полученные заранее эфиры енолов. Обсудим это отдельно.
Итак, в кислотно катализируемой альдольной конденсации равновесный енол реагирует с протонированным карбонильным соединением в присутствии сильной кислоты. Кислотно-катализируемая альдольная конденсация почти не применяется в современной химии потому что она очень неселективна, ее почти невозможно выполнить в перекрестном варианте между разными карбонильными соединениями даже в тех случаях, конда нормально работает ненаправленная конденсация, катализируемая основаниями. В кислотно-катализируемой конденсации образуются смеси продуктов, и она почти никогда не останавливается на простом альдоле или продукте его дегидратации, а идет дальше. Иногда этим даже можно воспользоваться, как в самоконденсации ацетона и других метилкетонов в 1,3,5-тризамещенные бензолы, но в большинстве случаев получается просто неопределенная смесь.
Некоторое минимальное применение кислотно-катализируемая конденсация находит для получения продуктов самоконденсации простых альдегидов и кетонов, при этом почти всегда получается продукт дегидратации. Например, имено так можно получить собственно кротоновый альдегид и окись мезитила (старинное название для продукта дегидратации самоальдоля из ацетона). Распишем реакцию на примере кротонового альдегида. Во-первых, обратим внимание на весьма примечательный факт: обе реагирующие частицы – электрофил и нуклеофил, карбонильная и метиленовая компоненты, протонированный карбонил и енол – образуются в результате действия одного и того же катализа фактически в одном и том же равновесии. Кислотный катализ умудряется добывать сразу оба типа реакционной способности, в отличие от основного катализа, который активирует только нуклеофильность. К сожалению, в химии такая доблесть не всегда ценится по достоинству: вот если бы на каждую молекулу енола образовывалась бы ровно одна протонированная карбонильная группа, и они тут же и количественно бы, и необратимо бы реагировали, и продукт этой реакции тут же бы куда-нибудь девался бы, например сам собой складывался бы в чистую баночку с карсивой этикеткой – то цены бы не было такой промысловатости. Увы, в реальности получается каша из десятков сцепленных друг с другом равновесий, все реагируют со всеми, и добром такой пир реакций не заканчивается. Тем не менее, наши далекие предки потратили много времени и сил на то, чтобы отработать методики таких простых реакций – температуру, концентрации, время, растворители, порядки смешения, скорость смешения и др. и пр. – и для таких реакций можно найти старые работающие методики, которые придется воспроизводить буквально и как можно ближе к тексту. Это занудно, но в целом дешево и работает, но по методике, скажем, для ацетальдегида не получится сконденсировать даже его ближайший аналог, для которого придется искать свою методику.

А что такое вообще таутомерия
Строго факультативно. Не читайте этот раздел, если не хотите забивать голову лишними сведениями, сеющими сомнения.
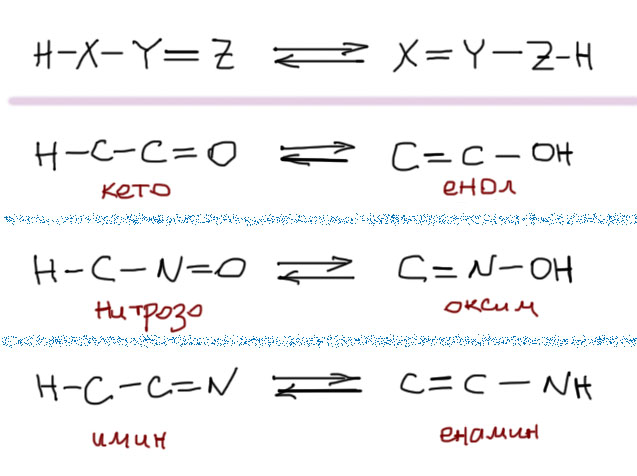
Таутомерия – это очень важная разновидность изомерии, в которой изомеры (таутомеры) связаны реальным равновесием, а превращение одного таутомера в другой происходит за счет переноса протона (такая таутомерия называется прототропной) или реже других атомов, например, металлов (тогда это металлотропная таутомерия) между атомами, связанными чередующимися простыми и кратными связями с одновременным перераспределением этих связей. Наиболее часто такой процесс происходит в трехатомных фрагментах (тогда таутомерия называется триадной), но бывает и таутомерия в пятиатомных, семиатомных и т.п. фрагментах, но это все довольно большие редкости, поэтому сразу ограничим себя только самой важной и невероятно часто встречающейся триадной таутомерией, в которой протон перемещается между крайними атомами трехатомного фрагмента. Примеров такой таутомерии многие десятки, вот хотя бы три примера (неиспользованные валентности не показаны) – кето-енольная, нитрозо-оксимная, имин-енаминная.
Некоторым читателям может прийти в голову другая аналогия – а как связана триадная таутомерия и аллильная перегруппировка, с которой мы тоже неплохо знакомы, хотя бы в такой форме как взаимопревращение продуктов 1,2- и 1,4-присоединения электрофилов к сопряженным диенам. Некоторым может даже прийти в голову такая вещь, как сигматропные перегруппировки, в частности чрезвычайно похожая на обычную таутомерию [1,3]-сигматропная перегрупировка.

Как ответить на этот вопрос? Увы, есть проблема. Понятие таутомерии не поддается формальному определению. Это частая ситуация в химии и вообще в науках, когда одна часть исследователей любит все обобщать и везде искать аналогии, а другая безуспешно стремиться к строгости формулировок, стараясь наоборот каждое определение делать максимально точным. Эти партии никогда не могут договориться, а поскольку в химии и вообще в науках (кроме марксизма-ленинизма) нет верховных начальников или жрецов, то все эти споры обычно ничем не кончаются. В химии своеобразным законодательным правом обладает уже знакомый нам ИЮПАК. По крайней мере, с номенклатурой органических, неорганических и координационных соединений и некоторыми другими вещами у этой международной организации что-то получилось – в научных статьях используют именно ее. ИЮПАК попробовал определить и наиболее важные термины химии и издал Золотую Книгу (IUPAC Golden Book, по цвету обложки вполне в геральдическом смысле, она не золотая, а всего лишь желтая, но именно так – золотом – называется этот цвет в гербах и флагах), или полностью – Компендиум Химической Терминологии. Золотую книгу можно с полными основаниями назвать эпическим провалом – о ее существовании мало кто знает, и почти никто ей не пользуется, но другого авторитетного источника терминологии все равно нет. Книга не смогла дать строгого определения и таутомерии, и это и есть надежный признак того, что такого определения дать и не получится. При этом, данное Золотой книгой определение не лишено простоты и изящества, что не удивительно, так как над определениями этой книги работали очень серьезные ученые. Согласно Золотой книге таутомерией является взаимопревращение изомеров, происходящее за счет перенос какого либо атома или остатка (G) с одновременной перестройкой связей, причем этот процесс должен а) происходить легко (readily) – и никто не объясняет, что значит “легко”; б) перемещающаяся группа или атом в процессе становится либо электрофугом (то есть перемещается, не забрав с собой электронную пару бывшей связи), либо нуклеофугом (то есть перемещается с парой электронов).
 Скажем ИЮПАК спасибо, потому что под это определение полностью подпадают и аллильные перегруппировки, в том числе и превращение продуктов 1,2-присоединения электрофилов к сопряженным диенам в продукты 1,4-присоединения, именно потому что определение Золотой книги допускает перемещение нуклеофуга, в частности, бромид-аниона вместо обычного для традиционного понимания таутомерии электрофуга типа протона. Тем не менее, вы практически никогда не встретите применения термина таутомерия к аллильным перегруппировкам. Этот пример показывает, насколько нестрогой является общепринятая химическая терминология, и как важно понимать суть происходящих процессов, а не уповать на механическое запоминание названий и правил.
Скажем ИЮПАК спасибо, потому что под это определение полностью подпадают и аллильные перегруппировки, в том числе и превращение продуктов 1,2-присоединения электрофилов к сопряженным диенам в продукты 1,4-присоединения, именно потому что определение Золотой книги допускает перемещение нуклеофуга, в частности, бромид-аниона вместо обычного для традиционного понимания таутомерии электрофуга типа протона. Тем не менее, вы практически никогда не встретите применения термина таутомерия к аллильным перегруппировкам. Этот пример показывает, насколько нестрогой является общепринятая химическая терминология, и как важно понимать суть происходящих процессов, а не уповать на механическое запоминание названий и правил.


Енолят-анионы
Раздел в работе. Пока только несколько важных вещей.
Енолят – это сопряженное основание енолизуемого карбонильного соединения. Обе таутомерные формы, енол и кето-форма, дают один и тот же енолят. Несимметричные кетоны, имеющие α-атомы углерода с обоих сторон от карбонильной группы, могут давать два разных енолята, и с этим связана проблема региоселективности в реакциях, идущих через еноляты – могут образоваться два разных продукта, а если еще учесть стереохимию, то даже больше. Стереохимию мы учитывать не будем.
Главная проблема, которая может и даже должна возникнуть при обсуждении реакций енолята, – почему он реагирует по атому углерода. Источник противоречия прост: с одной стороны мы весьма категорично говорим, что структура енолята почти точно отображается именно енолятной формой с отрицательным зарядом на кислороде. Следовательно, отрицательный заряд в еноляте и находится на атоме кислорода. Ну и что? Но разве нуклеофильность не связана именно с отрицательным зарядом на атоме? разве мы не утверждали, когда обсуждали нуклеофильность, что анионы – более сильные нуклеофилы, чем нейтральные молекулы?
Нет, конечно. Анионы – более сильные нуклеофилы только по сравнению с нейтральными молекулами, являющимися сопряженными кислотами этих анионов. Гидроксид-ион сильнее воды, а ацетат – уксусной кислоты, мы уже не говорим о разнице в нуклеофильности метильного карбаниона и метана. Да, и енолят – более сильный нукелофил чем сам енол, и это точно верное утверждение. Но мы не можем сравнивать два разных нуклеофила или, как в еноляте, два разных нуклеофильных центра только на основании заряда на атомах. Нет такой закономерности в представлениях о нуклеофильности, просто нет и все, а если вам кажется, что есть, то, уверяю вас, вы ее сами выдумали и сами же в нее и поверили.
А как же быть, какая есть закономерность? Вот мы точно в разделе о нуклеофильности связывали нуклеофильность и основность для элементов 2 периода, то есть как раз тех самых C, N, O. Больше основность (больше pK) – больше нуклеофильность. Это качественная тенденция, не дающая численных оценок, а работающая в терминах больше-меньше. Если разница большая, то и оценка получается довольно надежная. Да, вот это уже неплохо, ведь мы только что обсуждали, что само явление таутомерии связано с различием CH- и OH-кислотности кето-формы и енола, а это вполне можно интерпретировать так, что енолят простых карбонильных соединений – существенно более сильное основание Бренстеда-Лоури по атому углерода (этому кислотно-основному равновесию соответствует CH-кислотность кето-формы), чем по атому кислорода (этому равновесию соответствует OH-кислотность енола). Вроде работает.
Но некоторое ощущение неудовлетворености от такого объяснения не проходит. Если чуть-чуть забежать вперед, то там окажется, что еноляты 1,3-дикетонов и кетоэфиров типа ацетоуксусного эфира, тоже в основном работают как C-нуклеофилы, хотя там как раз кислотность кето-формы очень сильно повышена, и порядок основности по углероду и по кислороду совсем иной. Строго говоря, разница в основностях там не очень велика, и закономерность основность-нуклеофильность просто перестает работать, именно потому что это очень грубая, качественная закономерность.

Итак, ЖМКО работает. И можно на этом остановиться, если только не задавать вопроса, а почему. Это больше похоже не на теорию, а на банальное обобщение экспериментального материала. Это так и есть, но чтобы под это почти религиозное учение подложить что-то, напоминающее настоящую науку, не обойтись без квантовой химии, с которой мы совсем не в ладах. Иными словами, дальше можно не читать. Объяснения с точки зрения ЖМКО всегда принимаются, и на экзаменах, и даже в научной литературе.
Но можно и почитать. Самое популярное квантово-химическое описание того, как реагируют молекулы, использует теорию молекулярных орбиталей, и в ней сосредоточивает внимание не на всех орбиталях, а только на так называемых граничных, особенно двух из них – самой последней по энергии (считая снизу вверх) занятой – высшей занятой (ВЗМО), и самой первой незанятой – низшей свободной (НСМО). Когда говорят о реакции нуклеофил-электрофил, то довольно очевидно, что для нуклеофила мы берем его ВЗМО (нуклеофильность определяется парой электронов, которая пойдет на образование новой связи), а для электрофила – его НСМО (пустую орбиталь, ведь и электрофил всегда предоставляет пустую орбиталь для образования новой связи). Дальше, мы должны откуда-то взять эти орбитали (посчитать или просто оценить на глазок, если мы знаем, как это делается), и в этом случае направление реакции будет соответствовать максимальному взаимодействию этих граничных орбиталей, в свою очередь соответствующему взаимодействию между теми атомами, на которых в этих орбиталях вклады соответствующих атомов максимальны (молекулярные орбитали строятся из атомных, и каждый атом дает какой-то вклад, какие-то атомы больший, какие-то меньший или совсем незначительный). Займемся этим для реакции енолята. В этом случае, нам потребуется ВЗМО енолята, который даст нам представление о том, где находится больший вклад в эту орбиталь – на кислороде или на углероде. Электрофилом и его НСМО нам заниматься не придется, потому что там никаких сюрпризов быть не может, если у электрофила один центр, то на нем и нужная орбиталь.

Такой подход позволяет понять, почему два реагента реагируют именно по таким атомам, а не другим. Реагенты стараются сблизиться и образовать связи так, чтобы были максимальными орбитальные взаимодействия, среди которых считаются самыми главными взаимодействия ВЗМО-НСМО. Такой способ взаимодействия называют орбитальным контролем. И тогда, если немного подумать и сопоставить, именно орбитальный контроль соответствует взаимодействию мягкий-мягкий в ЖМКО. Фактически получается, что представление об орбитальном контроле это объяснение тому, как работает концепция ЖМКО. Мы даже можем сказать, что эта теория примиряет нас с теорией ЖМКО, которая сама по себе весьма голословна, но с подкладкой из орбитального контроля становится как будто бы обоснованной, по крайней мере в части мягкое-к-мягкому.
А тогда что такое взаимодействие жесткий-жесткий из ЖМКО? Это называется обычно зарядовым контролем, то есть таким направлением реакции, в которой максимальными делаются не орбитальные взаимодействия, а просто взаимное притяжение мест с максимальными зарядами. В протонировании енолята сильной кислотой, например, положительный протон предпочитает атом с наибольшим отрицательным зарядом – кислород. Но и заряды на атомах тоже берутся из квантово-химических расчетов и представлений, только в определении заряда участвуют не граничные орбитали, а все вообще орбитали, образованные атомными орбиталями валентной оболочки.
Вот как-то так приблизительно и принято объяснять реакции в последние лет 40-50 после первых работ Роберта Вудварда и Роалда Хоффмана в середине 1960-х. Это не единственный способ и не самый современный, но точно самый популярный, отмеченный Нобелевской премией 1981 года, сочетающий достаточно солидное научное обоснование, и вполне доступную нормальным людям наглядность.
Почему алкилируют енамины, а не еноляты
Вообще-то, если поискать в литературе, то реакции алкилирования енолятов вполне нередко попадаются. Да и в учебниках они упоминаются, часто вообще без комментариев. В реальных синтезах из литературы это почти всегда очень специфические случаи, а идея запоминать частные случаи в органической химии – плохая идея, слишком много в органике частных случаев, которые нельзя обобщить. Мы предпочитаем использовать более надежные методы, которые можно использовать для многих похожих реакций. И в случае выбора – енолят или енамин – в качестве нуклеофила для алкилирования, выбирать мы будем енамин.
Главная проблема при алкилировании енолятов простая – алкилирование сразу дает готовое карбонильное соединение, альдегид или кетон, а поскольку реакция алкилирования не очень быстрая, то в реакционной смеси одновременно оказываются и карбонильное соединение и непрореагировавший енолят. И они вступают – не могут не вступать – в реакцию альдольной конденсации алкилированного карбонильного соединения (в качестве карбонильной компоненты) и еще не прореагировавшего енолята (в качестве метиленовой компоненты). Результатом будут невысокие выходы продуктов алкилирования и образование смеси побочных продуктов.

Эта проблема особенно важна при использовании енолятов альдегидов: из-за очень высокой реакционной способности альдегидов альдольная конденсация пойдет с большой скоростью, и побочная реакция станет основной.
В отличие от енолятов, енамины при алкилировании непосредственно в реакции дают соли иминия, которые не реагируют с непрореагировавшим енамином (могли бы, но не хватает реакционной способности), поэтому алкилирование протекает намного более гладко, и всячески поэтому рекомендуется, хотя, как мы знаем, и у него есть проблемы – невысокая нуклеофильность енамина делает реакцию алкилирования медленной, если только не используются очень активные SN2-субстраты.